遗传标记(Genetic markers)是指与目标性状紧密联锁,同该性状共同分离且易于识别的可遗传的等位基因变异。目前,应用较为广泛的遗传标记有形态标记、细胞标记、生化标记和分子标记。DNA分子标记具有标记数量多、分布广、遍及整个基因组、多态性高、遗传稳定、不受环境限制的特点而成为一种重要的遗传标记技术,广泛应用于遗传育种、基因组作图、基因定位与克隆、遗传多样性检测、系统进化、分子分类与物种亲缘关系鉴别等方面(Patrick et al., 1997; Sasaki and Burr, 2000; Qian et al., 2001; Jeung et al., 2007; 王云生等, 2007)。目前常用几种标记如RFLP、AFLP、RAPD、SSR、ISSR等(周延清等, 2008)。这些技术应用有部分优点,也有一定局限性。如RFLP技术需要利用放射性同位素,实验成本较高;RAPD技术稳定性不太理想;SSR技术开发需要一定的DNA序列背景信息,引物开发难度大;ISSR由于是随机引物筛选,费事费力;AFLP由于得到的是酶切混合片段,不能将不同来源的相同长度大小DNA片段分开,技术相对比较粗放等。这些技术离理想的遗传标记技术还有一段差距,而且这些技术都基于毛细管电泳或凝胶电泳技术,整个检测过程自动化程度低,通量低,产出率低,精度低。
基因芯片技术是近年发展起来的一项新的生物技术(陈忠斌等, 2005)。芯片技术是将得到的探针DNA用芯片点样仪点于芯片载体上,然后将待检样品和芯片杂交,最后利用检测仪判断杂交信号大小。芯片样品点直径通常在25~500 μm,通常1 cm2可点样2 500个点阵,一般1张载玻片可容纳30 000左右DNA探针,芯片技术具有明显的高通量的特点,而且芯片技术是基于分子杂交的检测技术,与常规凝胶电泳技术相比具有高精度的特点,另外检测样品需求微量化。基因芯片技术已经在生命科学的许多领域得到广泛应用,如在医药、动植物疾病诊断、遗传代谢分析、食品安全检测和侦测等方面都有应用(陈忠斌等, 2005)。目前制作的基因芯片绝大部分属表达谱芯片,芯片点样的DNA主要是cDNA或合人工成的DNA,芯片检测的目的主要集中在新基因发现、基因功能研究、基因诊断、DNA序列分析、SNP检测等研究方面(梁秀彬等, 2002)。如果能将芯片技术的自动化、高通量、高精度特点和遗传标记技术结合起来,将基因组DNA制作成遗传标记的检测芯片,用于遗传分类、进化、遗传作图、辅助育种和遗传多样性检测等方面,必将极大促进遗传标记技术的发展。但基因芯片技术与遗传标记技术如何结合,如何应用,应用效果如何等问题,目前国内外在此方面应用研究报道较少,仅限于位于澳大利亚的Diversity Arrays Technology (DArT)技术实验室开展相关研究(Jaccoud et al., 2001; Wenzl et al., 2004; Xia et al., 2005; Wittenberg et al., 2005; Akbari et al., 2006; Yang et al., 2006)。
分布在我国南方的5种冷杉植物百山祖冷杉(A. besganzuensis) (吴鸣翔定名发表)、元宝山冷杉(A. yuanpaoshanensis) (吕庸浚和傅立国定名发表)、资源冷杉(A. ziyuanensis) (傅立国和莫新礼定名发表)、梵净山冷杉(A. fanjinshanensis) (黄威廉定名发表)和大院冷杉(A. dayuanensis) (刘起衔定名发表)均为我国的一级重点保护植物(《国家重点保护野生植物名录》)和濒危植物(《中国植物红皮书》),其中百山祖冷杉目前野生植株仅剩3 棵,被世界保护联盟维护物种生存委员会(SSCT)列为世界最濒危的12种植物之一。这5种冷杉植物的发现否定了以往亚热带的低、中山无冷杉分布的观点,被认为是植物地理学和植物学上的一大发现。由于冷杉属植物的分布受第四纪冰川时期的影响,因此上述冷杉植物是研究我国第四纪冰川时期植物区系和气候变迁的 “活化石”,对研究古气候、古地理和植物区系具有重要意义。目前这5种冷杉分类存在争议,现有分类的种内和种间遗传分化、遗传多样性不明,群体保护地位重要性不清。要开展有关研究,必须采用分子标记技术分析多态位点。多态位点数量具有重要作用,位点少,信息可靠性低,位点多可靠性高。多态位点的获得和检测样本大小及检测技术有关。在有关冷杉研究中遇到特别是百山祖冷杉,现有植株太少,满足不了一般群体取样样本数,常规凝胶电泳技术位点检测能力低,通量低,基于目前状况作者拟建立遗传标记芯片体系用于冷杉保护遗传学研究。
该技术原理和基本步骤为:首先提取高质量的基因组DNA,然后将基因组 DNA通过限制性内切酶酶切,产生不同大小的酶切片段,接着将酶切片段通过添加选择性碱基引物扩增,降低酶切片段总数量,再将这些选择性扩增后的酶切片段通过TOPO载体克隆,很多个克隆点构成一个芯片文库。此部分操作为芯片文库构建,不同的材料采用不同的酶切和引物扩增方法可以得到不同的芯片文库。克隆产物通过培养,PCR扩增,扩增后的DNA再经过纯化,最后将该纯化的克隆DNA点样在生物芯片上,制成一张用于遗传标记检测的基因组DNA芯片。当要利用该芯片对待测样品检测时,先将被检材料酶切,酶切后的PCR扩增物采用荧光标记,然后将标记物和芯片杂交,最后通过杂交信号检测判断检测样品与标记点同源性差异,杂交信号强,则同源性高,否则就低。
由于不同材料的基因组DNA复杂性不同,以及限制性酶切位点也存在很大差异,因此用不同的限制性酶切,以及采用不同添加选择性碱基引物扩增时产生的DNA片段大小、数量、以及片段在基因组上分布位置的均匀性都有差异,最后得到的有效标记数也不同。要得到高质量的基因组DNA芯片和得到符合一定质量参数的有效标记数,就需要研究合适DNA提取方法、合适的酶切和引物组合试验,笔者根据此思路开展了基因组DNA提取、多种不同酶切组合和不同引物扩增实验,成功制作了用于冷杉遗传多样性检测的芯片,并做初步应用分析。
1结果与分析
1.1冷杉基因组DNA提取
利用改良的CTAB法从冷杉叶片中提取冷杉基因组DNA,经检测样品D260 nm和D280 nm光度值,其比值都在1.9左右;风干后DNA溶液清亮,无黏稠感,实验结果说明提取的DNA纯度较好,样品中基本无多糖、蛋白质、和酚类等杂质。
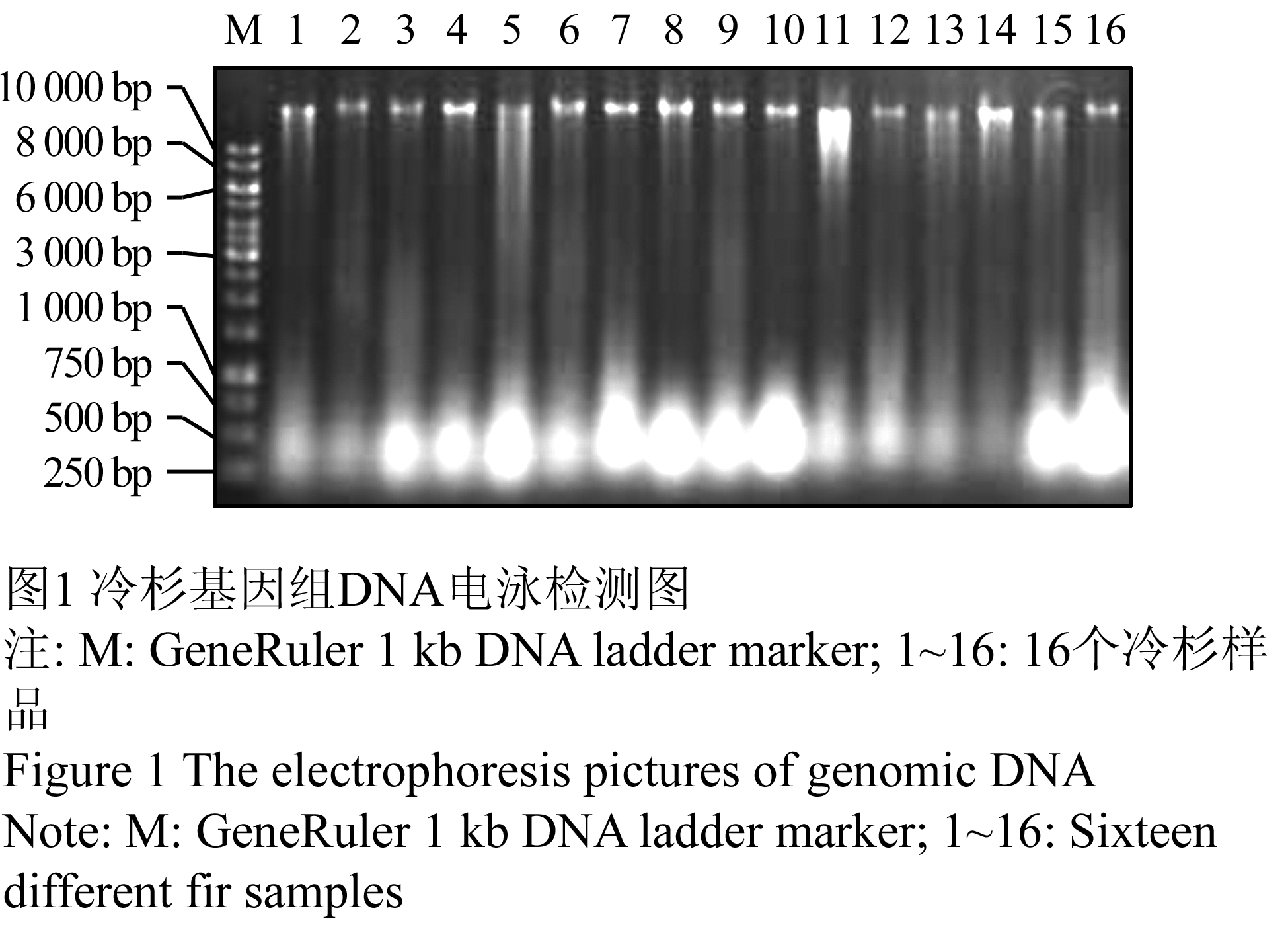
将提取的冷杉叶片DNA与生物芯片DNA工作缓冲液混合,室温静置3 h,利用琼脂糖凝胶电泳检测分析。如果DNA提取质量不好,或不适合DNA芯片工作缓冲液环境,则凝胶电泳后DNA可能检测不出或裂解。本实验检测结果如图1所示。由图可以看出,电泳样品孔均不发亮,各泳道DNA主带清晰,基本无弥散带,说明提取的DNA完整性较好,纯度也较高。电泳图谱显示有少许RNA 存在,但RNA在本实验系统中不影响实验结果。

图1 冷杉基因组DNA电泳检测图
Figure 1 The electrophoresis pictures of genomic DNA |
1.2冷杉基因组DNA不同组合酶切和不同引物扩增效果
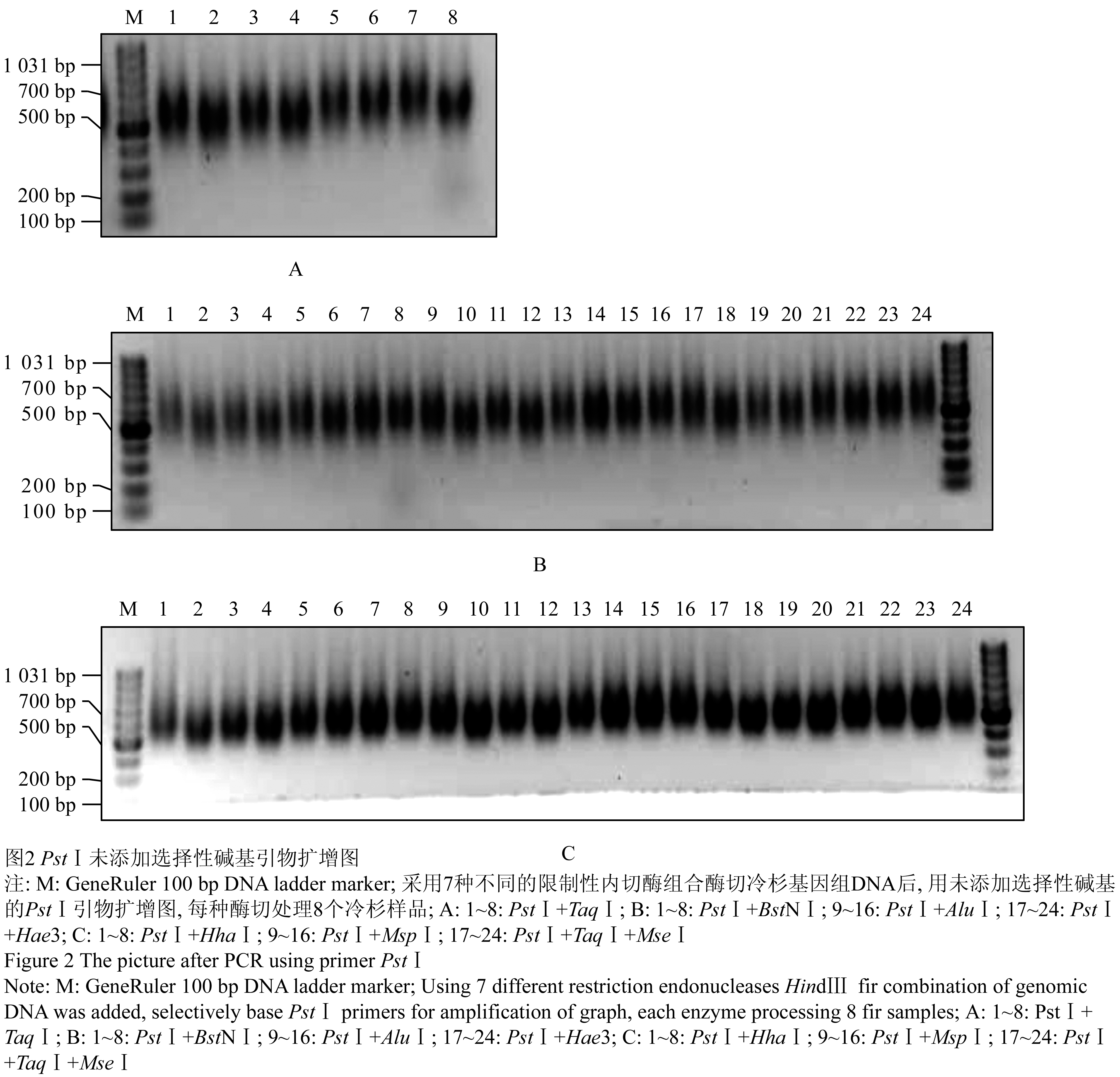
采用7种不同的限制性内切酶组合酶切冷杉基因组DNA后,用无选择性碱基PstⅠ引物扩增,扩增电泳图如图2,用添加选择性碱基CAG的PstⅠ引物扩增图如图3。从图2中可看出,酶切组合PstⅠ+TaqⅠ+MseⅠ (图2C, 泳道17~24)酶切片段大小分布在750 bp左右,而且分布比较均匀集中,分子量比较大的片段和比较小的片段相对较少。从该酶切组合的PstⅠ引物添加选择性碱基CAG扩增图3分析,所在泳道13、泳道14没有明显的条带,说明该酶切组合能较均匀切开冷杉基因组DNA。综合图2和图3分析认为PstⅠ+TaqⅠ+MseⅠ酶切组合效果最好。PstⅠ+BstNⅠ、PstⅠ+TaqⅠ的酶切效果次之,其它几种酶切组合不甚理想,表现为片段过大或均匀度差,出现明显条带。

图2 PstⅠ未添加选择性碱基引物扩增图
Figure 2 The picture after PCR using primer PstⅠ |
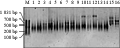
图3 PstⅠ添加选择性碱基CAG引物扩增图
Figure 3 The picture after PCR using primer PstⅠ+CAG and seven different combination of incision enzyme |
1.3冷杉芯片文库盘质量分析
基因组DNA经过酶切扩增后采用TOPO载体克隆,多个克隆构成芯片文库。在文库构建克隆挑选过程中,理论应该在文库盘每格(孔)位置上有一个克隆点,实际操作过程中有可能漏放或重放或挑选了空克隆,这样可能会影响芯片质量。为了保证文库盘质量,挑选的克隆点需要先检测,一般文库盘中错误克隆点必须要小于 5%,最好控制在2%以内水平。本实验文库盘克隆点检测部分电泳图样如图4示,从左致右1~33泳道,第1~16泳道为16个克隆点,第17泳道为间隔,第18~33为另外16个克隆点。从此电泳图可看出,挑选出的克隆片段大小主要约在500~1 000 bp左右,少数克隆在1 000~1 500 bp,而且每检测孔道均有1个克隆,满足芯片文库盘的质量要求。

图4 文库构建时克隆检测图
Figure 4 Testing clone for creating library |
1.4冷杉芯片质量分析
当芯片文库盘作好后,就可以将文库盘上克隆点点样于芯片上,制作成遗传标记检测芯片。芯片上样点的均匀程度是衡量芯片质量的重要指标。制作出一批芯片后,将采用部分样品和芯片杂交,通过杂交信号及参数可以判断芯片质量。
从表1可知,各张芯片的样点基本比率、中值、均值比率相关系数为0.866~0.976,说明样点点样及杂交后的均匀性很好,质量可靠。信噪比是判断检测系统性能和芯片杂交稳定性的重要参数,理想检测仪器的信噪比都在一定范围。从表1中可看出参考通道信噪比为79.017~92.024,杂交后检测通道信噪比为20.786~32.827,符合检测系统的理想信噪比值。综合样点的3种比率相关系数及信噪比参数可以判断,采用该方法得到的芯片质量比较好,可以用于有关冷杉检测分析工作。

表1 冷杉遗传标记芯片质量参数表
Table 1 The table of qualitative parameters of microarray slides |
1.5有效标记得率分析
本试验测试中共随机挑选有效克隆4 608个点样制作遗传标记检测芯片,经8样品杂交检测数据分析后得到重复性100%、召回率85%、P值大于75%、Q值大于75%的标记点共821个,有效标记点约17.8%。
1.6 8样品821个标记位点遗传多样性及遗传聚类分析
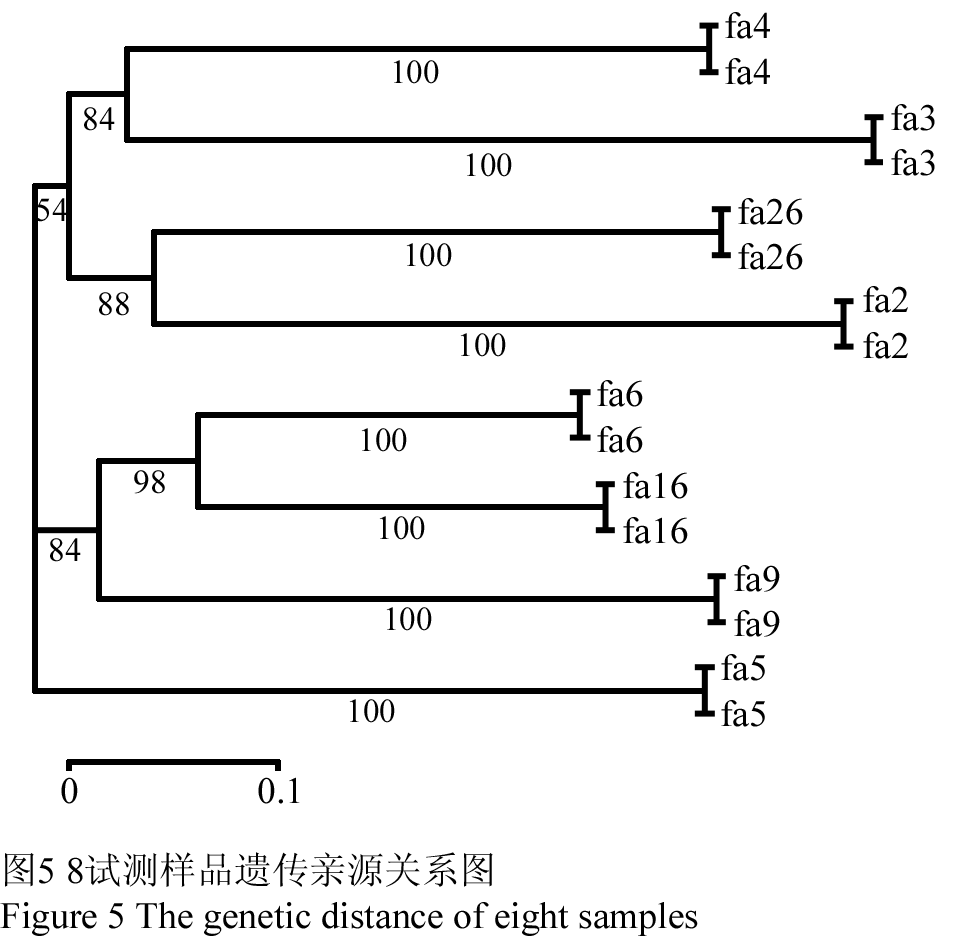
将8个不同的样品,按上述方法经杂交检测后取标记参数值均为P值、Q值均大于75%,重复性为100%,召回率大于85%的标记点统计分析。分析得到标记点的多态性位点百分比率P为100%,8样品多态性信息含量指数为0.4。采用DARwin5软件对8样品聚类分析,如图5所示。用该种方法可以有效分析样品间的遗传距离或亲缘关系,而且用不同荧光标记的同一重复样品亲缘关系聚类支持率为100%,说明该种方法切实可行,而且重复性,可靠性均较理想。
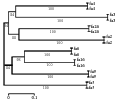
图5 8试测样品遗传亲源关系图
Figure 5 The genetic distance of eight samples |
2讨论
遗传标记技术广泛应用于分子育种、图谱构建、基因定位与克隆、物种分类与进化、多样性检测等。目前常用的检测技术还都基于凝胶电泳技术,具有产出率低、精度低、通量低的共性。基因芯片技术具有检测系统微型化、样品需求的微量化、检测的高效化及高通量化的优势和特点,从本实验结果看,芯片技术可以很好的和标记技术结合起来,充分发挥芯片技术高通量、高精度特点以及标记技术应用广泛的特点。利用此技术具有明显的优点:(1)标记源于基因组DNA某部分未知片段,不依赖DNA背景信息,即使是一个未知物种也可开展工作,该优势在许多遗传负荷大,前期遗传背景研究较少的濒危木本植物多样性研究中尤其重要。(2)高通量,标记数多、检测效率高。通常一张芯片可容纳3×104~4×104左右标记,而且样品处理也可采用批量式,检测效率很高,常规凝交电泳标记无法达到的。(3)利用该标记技术产生的标记数几乎可无限量扩展,直至覆盖整个基因组,而且要想增加标记数也比较容易;标记数越多,遗传多样性检测精度越高。(4)得到的文库具有一定通用性,一旦建成一个比较好的文库后,相近物种的样品检测将变得非常容易。该技术非常适合并未完成全基因组测序的物种开展分类进化、辅助育种、图谱构建、种质真假鉴定等研究,应用潜力广泛。
本试验开展了冷杉遗传标记芯片技术研究,从各项芯片质量、检测参数和位点参数数据分析,得到的芯片遗传标记芯片满足质量要求,有效标记位点约17.8%,利用通过此方法得到的标记分析标记点的多态性位点百分比率为P为100%,8个样品Nei基因遗传多样性指数为0.4,而且样品间遗传距离的支持率以及重复样品的位置关系都比较理想,说明该种方法切实可行,重复性,可靠性均较理想,可以应用于冷杉遗传分类、遗传多样性检测中。
本文进行的8个冷杉样品有关分析只是为了供遗传标记芯片质量检测和分析试用,8个样品并不能较好代表某冷杉群体遗传多样性水平,冷杉群体间分类和遗传分化问题探讨另文待发(饶龙兵, 2009)。
DArT技术实验室已成功将DArT技术运用在大麦(Hordeum)、小麦(Triticum)、水稻(Oryza)、苹果(Malus)、木薯(Manihot)、拟南芥(Arabidopsis thaliana)、木豆(Cajanus)、高粱(Sorghum)、西红柿(Lycopersicon esculentum)、甘蔗(Saccharum)等的分子育种、亲源分析与种质鉴定、图谱构建、遗传多样性研究。
3材料与方法
3.1冷杉基因组DNA提取
材料:新鲜冷杉叶片,材料包含有元宝山冷杉(A. yuanpaoshanensis),取自广西元宝山;梵净山冷杉(A. fanjinshanensis),取自贵州梵净山;百山祖冷杉(A. besganzuensis),取自浙江庆元百山祖;资源冷杉(A. ziyuanensis),取自广西资源县、和大院冷杉(A. dayuanensis),取自湖南炎陵;以及日本冷杉(A. firma S.et),取自江西庐山等。除日本冷杉来源人工引种林外,其它几种冷杉均来自该种冷杉天然野生群体。
提取方法:①取粉末0.3 g;②在10 mL离心管中加3 mL EB,100 μL β-巯基乙醇,涡旋混匀后,在5 000 g离心力下离心10 min(室温),去上清液;③再加入1 mL EB,2 mL LB和200 μL 5% Sarkosyl,颠倒离心管,混匀;④ 60℃恒温水浴1.5 h,水浴期间摇动离心管3~4次;⑤加入24:1的氯仿:异戊醇3 mL,混匀静置10 min;再5 000 g室温离心10 min;⑥将上清液转于新的离心管中,重复操作步骤⑤、⑥一次;⑦向上清液中加入2倍体积的-20℃无水乙醇,然后轻轻颠倒离心管,混匀,静置15 min,自然沉淀,去上清。⑧向沉淀中加入2 mL -20℃ 70%乙醇,轻轻震荡,使DNA重悬于乙醇中。接着将重悬液转入2.5 mL离心管,500 rpm离心10 s,去上清。⑨最后将离心管斜放于离心架上,自然风干后加TE保存;实验中EB配方为(500 mL):ph 8.0 100 mmol/L Tris-Hcl;ph 8.0 50 mmol/L EDTA;灭菌,使用前加2.0 g亚硫酸氢钠,及10 g PVP,31.88 g山梨醇;LB配方为(500 mL):ph 8.0 200 mmol/L Tris-Hcl、ph 8.0 50mmol/L EDTA、58.45 g NaCl、15 g CTAB;TE配方为:ph 8.0 10 mmol/L Tris-Hcl、ph 8.0 1 mmol/L EDTA。
DNA纯度与完整性检测方法:基因组DNA纯度检测:DNA样品按30倍稀释后,在260 nm和280 nm处测定其紫外吸收值,计算出D260 nm/D280 nm的比值。基因组DNA的完整性检测:取2 μL DNA提取液,加3 μL上样缓冲液(内含0.25%溴酚蓝, 40%蔗糖),用0.7%琼脂糖凝胶(内含0.5 mg/L的EB(溴化乙锭))电泳,电泳缓冲液为1×TAE,电压110 V,电泳40 min后,紫外凝胶成像拍照(图1)。
3.2冷杉基因组DNA不同组合酶切和不同引物扩增效果
材料:上述几种冷杉基因组DNA。
方法:采用7种不同内酶切组合酶切基因组DNA。扩增引物1:采用PstⅠ酶切对应引物,未添加选择性碱基扩增,每种不同酶切组合处理8个冷杉样品,见电泳图2;扩增引物2:采用PstⅠ酶切对应引物再添加+CAG 3个选择性碱基,每种不同酶切组合处理2个冷杉样品,电泳图3。操作步骤:取冷杉基因组DNA 1 μL加入酶切反应混合体系进行酶切和接头连接。(酶切反应混合体系包括: PstⅠ及不同的酶切组合, PstⅠ人工接头, BSA, 酶切缓冲液, T4DNA连接酶, 去离子水)。酶切时间如下:1:PstⅠ+BstNⅠ先37℃ 2 h后60℃ 2 h;2:PstⅠ+TaqⅠ先37℃ 2 h后60℃ 2 h;3:PstⅠ+Hae3 37℃ 3 h;4:PstⅠ+AluⅠ 37℃ 3 h;5:PstⅠ+MspⅠ先37℃ 2 h后80℃ 20 min;6:PstⅠ+HhaⅠ先37℃ 2 h后80℃ 20 min;7:PstⅠ+TaqⅠ+MseⅠ 先37℃ 3 h再60℃ 2 h后65℃ 20min。酶切后产物添加dNTP、DNA聚合酶及PstⅠ对应的引物、PCR缓冲液,进行PCR扩增,扩增条件为94℃ 1 min、60℃ 40 s、72℃ 50 s共30个循环。扩增后的产物用1.2%凝胶电泳检测。
3.3基因组DNA芯片文库构建
芯片文库构建材料:芯片文库材料来源和库容大小可以根据不同研究目的构建不同大小的文库。如果建库材料来源比较广,而且库容比较大则可应用于多种材料的样品检测。本试验建库材料包含有元宝山冷杉(A. yuanpaoshanensis)、梵净山冷杉(A. fanjinshanensis)、百山祖冷杉(A. besganzuensis)、资源冷杉(A. ziyuanensis)、大院冷杉(A. dayuanensis),以及日本冷杉(A. firma S.et),主要目的是为以后相关材料检测准备。
芯片文库构建方法:基因组DNA提取采用前法。取上述建库材料各样品的基因组DNA 1 μL加入酶切反应混合体系,60℃温育90 min进行酶切和接头连接。酶切后的产物在扩增反应混合液中(包括PCR缓冲液, dNTP, DNA聚合酶及PstⅠ 引物)进行PCR扩增,扩增条件94℃ 1 min,60℃ 40 s,72℃ 50 s共30个循环。将不同来源(材料, 样品)的PCR扩增产物各取少量等量混合,然后利用TOPO-TA克隆法克隆。再将克隆转化物转到培养基中培养增殖,增殖后的克隆菌落在扩增反应液中直接 PCR扩增(扩增反应液包括: 扩增缓冲液, dNTP, DNA聚合酶及M13前向引物和M13后向引物)。扩增条件95℃ 4 min,57℃ 35 s,72℃ 1 min,共35个循环。PCR扩增后的产物烘箱干燥,然后再用乙醇洗涤两次,最后加入芯片点样缓冲液,利用芯片点样仪点样载玻片上,每样品4次重复,点样后的载玻片上经过DNA与基片高温再水合、紫外线交联固定及干燥处理,制成一批遗传标记检测芯片,置于芯片盒避光保存备用。
3.4试测样品处理及与芯片杂交
试测材料来源及处理方法:待检8份材料来源于梵净山冷杉野生群体。处理方法:提取样品基因组DNA,经PstⅠ+TaqⅠ+MseⅠ 限制性内切酶酶切,再采用和前面文库构建相同的方法PCR扩增、样品DNA沉淀、洗涤等处理。将处理过的样品DNA与荧光标记反应液混合标记样品DNA (荧光标记混合液包括dNTP, Klenow酶, 随机引物, 缓冲液)。每一待测样品均用两种不同荧光标记,作为标记重复。
芯片与试测样品的杂交及检测:将标记好的试测样品DNA和杂交缓冲液混合,再使样品DNA高温变性,变性好的混合液用吸头滴加到芯片上,盖上盖玻片,置于一定湿度和温度的杂交仪中,杂交12~20 h。杂交后的芯片采用杂交清洗液依次洗涤。洗涤后的杂交芯片用离心机甩干,再干燥,最后用芯片扫描仪扫描,进行数据分析。由于每个克隆在芯片上有4次重复,且每一待测样品均采用2种荧光标记,故实际上每个样品信号值有8次杂交重复。
芯片点样采用BioRobotics MicroGrid点样系统;芯片扫描采用Tecan LS Reloaded扫描系统。
3.5芯片数据分析
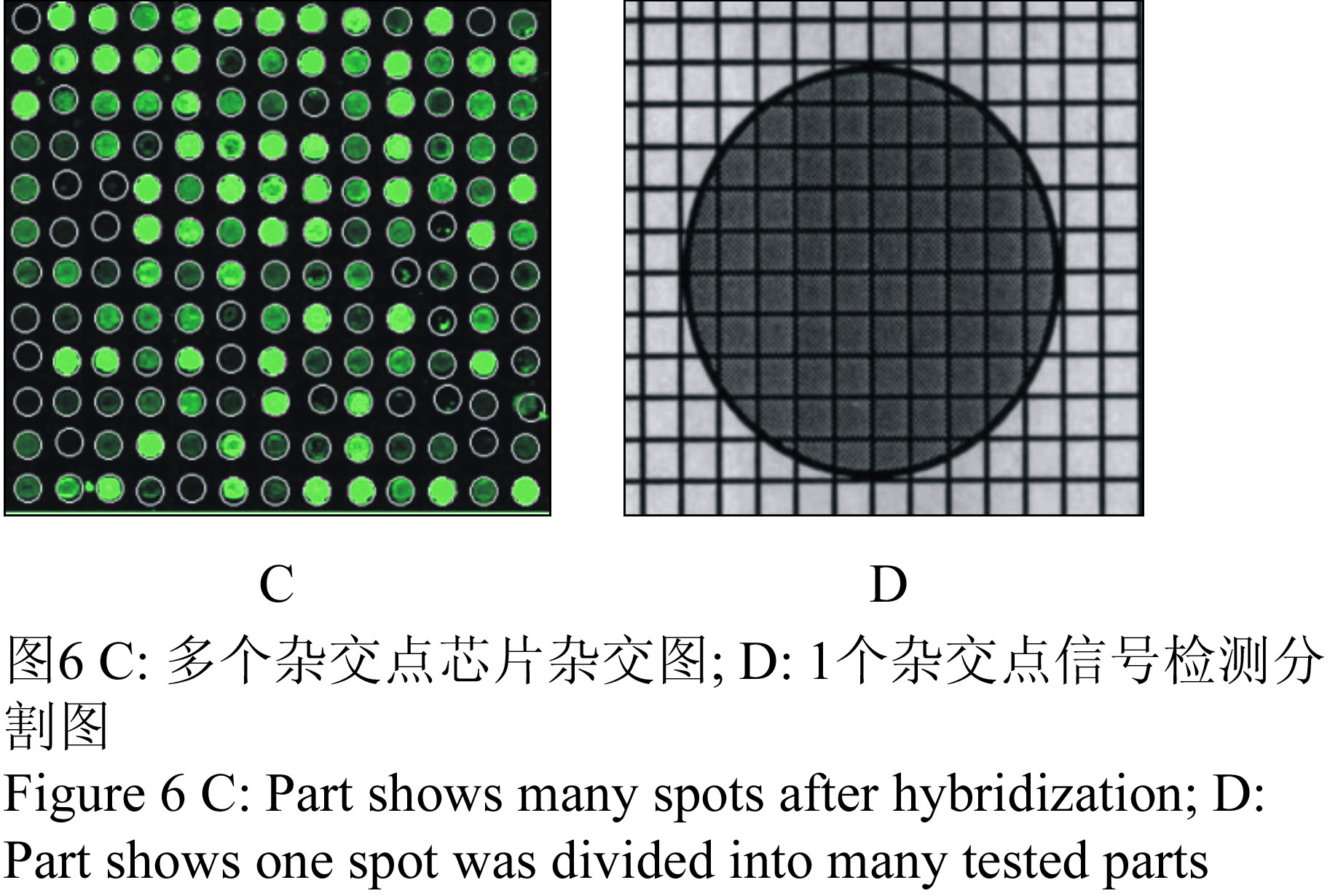
信号数据分析参数及计算方法如下:为了检测芯片点样质量和杂交信号质量,需要将每个杂交点样(图6C为多个杂交样点杂交信号实际照片),采用分割法再分成多个信号检测点(图6D所示)分析,每个信号检测点将有一个检测值,将这些被分割的检测点的检测值按照一定数学统计分析后将得到某杂交样点的信号值。芯片点样的质量和杂交信号质量可由以下参数判断:

图6 C: 多个杂交点芯片杂交图; D: 1个杂交点信号检测分割图
Figure 6 C: Part shows many spots after hybridization; D: Part shows one spot was divided into many tested parts |
①信号强度比率:定义为log(杂交信号强度-杂交背景强度/参考信号强度-参考背景强度);
②信号强度基本比率:指某一样点被分割成许多检测点后,所有信号切割位点的众数的中值;
③信号强度中数比率:指某一样点被分割成许多检测点后,所有信号切割位点的中位数比率。RatioMed=log(杂交信号强度中值-杂交背景强度中值/参考信号强度中值-参考背景强度中值);
④信号强度平均数比率(RatioAvg)指某一样点被分割成许多检测点后,所有信号切割位点的平均数比,RatioAvg=log(杂交信号强度平均数-杂交背景强度平均数/参考信号强度平均数-参考背景强度平均数);
⑤信噪比:A参考通道信噪比(SigToNoiseRef)=(参考通道强度-参考通道背景值)的中值/参考通道背景值标准差;B检测通道信噪比(SigToNoiseTarget)=(检测通道强度-检测通道背景值)的中值/参考通道背景值标准差。
多态位点分析参数及计算方法如下:在进行同一批芯片和多个样品杂交检测分析时,芯片上的标记点和不同的样品杂交会显现不同的杂交信号,将杂交点信号经数学分析后会出现1、0、-(不能判断) 3种判断结果。为了提高实验检测精度,必须设置同一位点和同一样品的多次杂交重复,结合召回率、P、Q值可判断某标记位点质量。
①重复性Reproducibility:重复性为判读结果相同次数占总重复次数的比率,或为每位点判读结果相同的概率。如果所有重复都出现判读结果相同,则重复性为100%。Repro=∑i=0 Nrep (N-difi)/Nrep (Nrep重复次数, difi为不同的次数);
② 召回率Call rate:召回率指某个位点上能判读得到的赋分值(1, 0)的样本数占总样本数的百分比。其相对值为该位点不能判读为(1, 0)的样本数(即缺失值样本数)占总样本数的比率。该值的高低代表该位点判读情况好坏的程度,可作为该位点是否选取的依据之一,如果该位点在多个样品中都不能判读出0、1结果,则该位点不被选取。Callrate=100×(∑i N scorei/N) (N总样本数, scorei能赋分的样本数);
③Q 值:Q值为某位点0、1两分后该位点强度比率中最低值组(0组)信号数值变异与强度比率最高值组(1组)信号数值变异之和占总变异的百分数。如果Q值越大说明分组后的方差占总方差比重越大,说明0、1分组效果越好。Q值的强度比率值选取可以是强度基本比率、中数比率和平均数比率三种中任1组值,因此Q值有 3种,取最大值作为该位点的Q值。Q=max(i×(x-xloi)²+(N-i)×(x-xhii)²)/N×var(x)×100 (0≤i<N, X为N个点的平均值, xloi为第i组最低值平均, xhii为第i组最高值组平均, var(x)代表N个点的方差)。
④P 值:P值和Q值类似,不同之处在于Q值分别统计3种强度比率中1种,然后取最大值。而P值同时考虑这3种强度比率,取3组中中心值比率。同样如果P值越大说明分组后的方差占总方差比重越大,0、1分组效果越好。P值是Q值的补充参数。P=(n1×(g-g1)²+n2×(g-g2)²/∑i=1 n1+n2 (xi-g)²×100 (n1和n2分别代表聚类到0, 1的点的个数; g是3个强度比率的中心值, g1和g2是n1和n2点中心值)。位点好坏和筛选参数的重要性程度依次为重复性、召回率、P值和Q值。
⑤有效标记数:指某标记位点满足一定域值(P值, Q值, 重复性, 召回率)的标记数。有效标记率指某标记位点的标记参数(P值, Q值, 重复性, 召回率)达到一定域值的标记数占总标记的百分数。
作者贡献
饶龙兵为本研究的具体完成人。
致谢
本研究由国家自然科学基金项目(30700644)资助。作者感谢澳大利亚Diversity Arrays Technology实验室Kilian A、Xia L.及其他几位同事在遗传标记芯片技术方面给予的支持和帮助,感谢百山祖自然保护区、元宝山自然保护区、资源冷杉保护区、梵净山自然保护区、桃源洞自然保护区在样品采集过程中给予的协助。
参考文献
Akbari M., Wenzl P., Caig V., Carling J., Xia L., Yang S., Uszynski G., Mohler V., Lehmensiek A., Kuchel H., Hayden M.J., Howes N., Sharp P., Vaughan P., Rathmell B., Huttner E., and Kilian A., 2006, Diversity Arrays Technology for high-throughput profiling of the hexaploid wheat genome, Theor. Appl. Genet., 113(8): 1409-1420 http://dx.doi.org/10.1007/s00122-006-0365-4
Chen Z.B., ed, 2005, Bio-chip Technology, Chemical Industry Press, Beijing, Chinese, pp.1-23 (陈忠斌, 2005, 生物芯片技术, 化学工业出版社, 北京, 中国, pp.1-23)
Jaccoud D., Peng K., Feinstein D., and Kilian A., 2001, Diversity Arays: a solid state technology for sequence information independent genotyping, Nucleic Acids Res., 29(4): E25 http://dx.doi.org/10.1093/nar/29.4.e25
Jeung J.U., Kim B.R., Cho Y.C., Han S.S., Moon H.P., Lee Y.T., and Jena K.K, 2007, A novel gene, Pi40(t), linked to the DNA markers derived from NBS-LRR motifs confers broad spectrum of blast resistance in rice, Theor. Appl. Genet., 115(8): 1163-1177 http://dx.doi.org/10.1007/s00122-007-0642-x
Liang X.B., Zhang H., Wang H.Y., Screening for genes related to novel gene AngRem l04 by gene chip, Zhonghua Shenzang Zazhi (China J. Nephrol.), 18(3): l59-165 (梁秀彬, 张宏, 王海燕, 2002, 采用基因表达谱芯片技术筛选新基因AngRem l04的功能相关基因, 中华肾脏病杂志, 18(3): l59-165)
Patrick J.C., Susan K.B., and Norman F.W., 1997, Random amplified polymorphic DNA -based genetic linkage maps of three cultivars, Amer. Hort. Sci., 122(3): 350-359
Qian W., Ge S., and Hong D.Y., 2001, Genetic variation within and among populations of a wild rice Oryza granulate from China detected by RAPD and ISSR marks, Theor. Appl. Genet., 102: 440-449 http://dx.doi.org/10.1007/s001220051665
Rao L.B., 2009, Study on conservation genetics of five endangered fir by micrroarray technology, Dissertation for doctorate,China Academy of Forestory, supervisor: Chen Y.T., pp1-96 (饶龙兵, 2009, 基于生物芯片标记技术的5种濒危冷杉保护遗传学研究, 中国林科院博士学位论文, 导师: 陈益泰, pp.1-96)
Sasaki T., and Burr B., 2000, International rice genome sequencing project: The effort to completely sequence the rice genome, Curr. Opin. Plant Biol., 3(2): 138-141 http://dx.doi.org/10.1016/S1369-5266(99)00047-3
Wang Y.S., Huang H.W., and Wang Y., 2007, Recent progress in plant molecular population genetics, Yichuan (Hereditas), 29(10): 1191-1198 (王云生, 黄宏文, 王瑛, 2007, 植物分子群体遗传学研究动态, 遗传, 29(10): 1191-1198)
Wenzl P., Carling J., Kudrna D., Jaccoud D., Huttner E., Kleinhofs A., and Kilian A., 2004, Diversity Arrays Technology (DArT) for whole-genome profiling of barley, Proc. Natl. Acad. Sci., 101(26): 9915-9920 http://dx.doi.org/10.1073/pnas.0401076101
Wittenberg A.H, van der Lee T., Cayla C., Kilian A., Visser R.G., Schouten H.J., 2005, Validation of high-throughput marker technology DArT using the model plant Arabidopsis thaliana, Mol. Genet. Genomics, 274(1): 30-39 http://dx.doi.org/10.1007/s00438-005-1145-6
Xia L., Peng K., Yang S.Y., de Vicente MC, Fregene M., and Kilian A., 2005, DArT for high-throughput genotyping of Cassava (Manihot esculenta) and its wild relatives, Theor. Appl. Genet., 110(6): 1092-1098 http://dx.doi.org/10.1007/s00122-005-1937-4
Yang S.Y., Pang W., Ash G., Harper J., Carling J., Wenzl P., Huttner E., Zong X.X., and Kilian A., 2006, Low level of genetic diversity in cultivated pigeonpea compared to its wild relatives is revealed by diversity arrays technology, Theor. Appl. Genet., 113(4): 585-595 http://dx.doi.org/10.1007/s00122-006-0317-z
Zhou Y.Q., Yang Q.X., Zhang G.N., (eds), 2008, Biologic genetic marker and application, Chemical Industry Press, Beijing China, pp.1-13 (周延清, 杨清香, 张改娜, 编著, 2008, 生物遗传标记与应用, 化学工业出版社, 中国, 北京, pp.1-13)

 作者
作者  通讯作者
通讯作者