研究论文
Cupriavidus campinensis BJ71菌株2, 4-D降解基因tfdA生物信息学分析 
2贵州大学生命科学学院山地植物保护与种质创新省部共建教 育部重点实验室, 贵阳, 550025;
3贵州大学贵州省农业生物工程重点实验室, 贵阳, 550025
 作者
作者  通讯作者
通讯作者
计算分子生物学, 2014 年, 第 3 卷, 第 5 篇
收稿日期: 2014年09月16日 接受日期: 2014年09月30日 发表日期: 2014年10月10日
引用格式(中文):
韩丽珍, 李翠翠, 赵德刚, 2014, Cupriavidus campinensis BJ71 菌株 2, 4-D 降解基因 tfdA 生物信息学分析, 基因组学与应用生物学, 33(3): 478-486 (doi: 10.13417/j.gab.033.000478)
引用格式(英文):
Han L.Z., Li C.C., and Zhao D.G., 2014, Bioinformatics Analysis of 2, 4-D-Degrading Gene tfdA of Cupriavidus campinensis BJ71, Jiyinzuxue Yu Yingyong Shengwuxue (Genomics and Applied Biology), 3(5): 1-11 (doi: 10.13417/j.gab.033.000478)
本文以2,4-二氯苯氧乙酸高效降解菌Cupriavidus campinensis BJ71菌株中克隆的tfdA基因全序列为研究对象,利用生物信息学方法分析该基因及其推测出的编码蛋白理化性质、蛋白的亲水性或疏水性及 亚细胞定位、预测信号肽、跨膜结构域、蛋白二级结构及模体、三级结构等特性。结果表明,BJ71菌株的 tfdA基因开放阅读框全长 864 bp,系统发育树表明其属于Ⅰ族tfdA基因。TfdA蛋白包含287个氨基酸,分子量为32 kD、理论等电点为6.01,是一种不含信号肽、定位于细胞质中的可溶性亲水蛋白。α螺旋和无规卷曲是该蛋白主要的二级结构元件。用RaptorX进行三级结构建模Ramachandran评估显示获得了合理可靠的TfdA蛋白结构模型。
高浓度2,4-二氯苯氧乙酸(2,4-D)作为一种防除阔叶杂草的选择性除草剂,自20世纪40年代工业化生产以来已被广泛应用了六十多年(韩丽珍和赵德刚, 2012)。尽管2,4-D的半衰期较短,但大量使用有可能在土壤中蓄积、通过径流进入地表水,甚至渗滤进入地下水中造成污染(Cox et al., 2000; Starrett et al., 2000; Johannesen and Aamand, 2003; Farenhorst et al., 2008)。环境中的很多微生物都可以降解或耐受2,4-D已经分离到的菌株基于进化和系统发育树被分为3群(Kamagata et al., 1997)。第1群包含在生长较快的β-和γ-变形菌纲中,基于tfdA基因的差异进一步分为3个族, Ⅰ族以澳大利亚分离到的Cupriavidus necator (原称为 Alcagenes eutrophus, Ralstonia eutropha, Wautersia eurtropha) JMP134菌株tfdA基因为代表,Ⅱ族以 Burkholderia sp. RASC菌株tfdA基因为代表,Ⅲ族以 Burkholderia cepacia 2a菌株tfdA基因为代表(McGowan et al., 1998);第2群和第3群含有tfdA-like基因,通常降解能力较弱(Kitagawa et al., 2002; Itoh et al., 2004; Zaprasis et al., 2010)。其中研究较为清楚的是C. necator JMP134 菌株的tfdA基因,编码产物为Fe(Ⅱ)/α-酮戊二酸依赖性双加氧酶TfdA,与来源于Escherichia coli牛磺酸双加氧酶TauD同属于第二群α-酮戊二酸依赖性双加氧酶家族成员(Hogan et al., 2000; Müller et al., 2006)。研究表明,TfdA蛋白在代谢2, 4-D为2, 4-二氯苯酚(2, 4-DCP)和乙醛酸的同时,α- 酮戊二酸被转变为琥珀酸(Fukumori and Hausinger, 1993)。Elkins 等(2002)利用E. coli牛磺酸双加氧酶TauD和肉桂酸合成酶CAS1的晶体结构(PDB ID: 1gqw, 1DS1)为模型预测了C. necator JMP134菌株TfdA蛋白的三级结构,但后期的研究表明来源于E. coli和Pseudo- monas putida的TauD并非二聚体结构,而是四聚体的晶体结构(Knauer et al., 2012)。
在前期研究中,我们从2, 4-D污染的麦田土壤中分离筛选到一株2, 4-D降解能力较强的Cupriavidus campinensis BJ71菌株,并克隆了存在于质粒上的2, 4-D降解基因tfdA全长序列,初步分析发现与C. necator JMP134菌株tfdA基因存在一定差异。本研究通过构建系统发育树对获得的tfdA基因进行分析,通过生物信息学方法对 tfdA 编码产物进行预测,可为进一步研究该基因的表达及突变奠定基础。
1 结果与分析
1.1 tfdA基因保守结构域分析及系统发育树构建
前期克隆的2,4-D高效降解菌C. campinensis BJ71菌株tfdA基因全长序列864 bp,通过Bioedit软件翻译成长度为287个氨基酸序列的蛋白质(图1)。CD Search分析表明tfdA基因编码产物为2, 4-D/α-酮戊二酸依赖性双加氧酶TfdA,具有TauD多重结构域,属于CAS类似的超家族(图2)。与目前报道的代表性的Ⅰ族、Ⅱ族、Ⅲ族tfdA基因以及tfdA-like基因、tauD基因共同构建系统发育树,结果可见,我们从BJ71菌株中克隆得到的2,4-D降解基因tfdA与A. eutrophus JMP134菌株tfdA聚于一枝,属于Ⅰ族 tfdA基因(图3)。该基因编码产物 TfdA蛋白与JMP134菌株TfdA相比,存在27个氨基酸差异(图 4)。
.png) 图 1 Cupriavidus campinensis BJ71 菌株 tfdA 基因及推导出的编码氨基酸序列 |
.png) 图 2 CD-Search 分析 C. campinensis BJ71 菌株 tfdA 基因保守结构域 |
.png) 图 3 MEGA5.0 构建 C. campinensis BJ71 菌株全长 tfdA 基因的系统发育树 |
.png) 图 4 ClustalW2 比对 BJ71 菌株 tfdA 基因编码蛋白与 C. necator JMP134 TfdA 的氨基酸序列 |
1.2 TfdA蛋白基本理化性质分析
通过ProtParam程序对其氨基酸组成及原子组成进行分析,预测蛋白分子量为32 kD,理论pI值为6.01,不稳定系数为50.68,高于40,其在E. coli中的降解半衰期大于10 h,属于不太稳定的蛋白。其氨基酸组成见表1,组成中含量较为丰富的氨基酸有3种,分别为Ala 33个(11.5%)、Arg 30个(10.5%)和Leu 28个(9.8%),含量最低的为Cys、仅有3个(1.0%);带正电荷(Arg+Lys)和负电荷(Asp+Glu)的氨基酸残基分别有34个和39个。脂肪族氨基酸指数较高,达82.65。
.png) 表 1 C. campinensis BJ71 菌株 TfdA 蛋白氨基酸组成 |
1.3 TfdA蛋白信号肽、跨膜区、定位及亲疏水性分析
信号肽由位于蛋白N端的可被剪切的15~30个氨基酸的前导序列组成,信号肽的预测有助于蛋白功能域的划分以及细胞定位;以D-cutoff 0.450,基于综合人工神经网络的方法,用SignalP 4.1预测信号肽结果表明,TfdA蛋白的信号肽原始剪切位点的最高得分值(Y-score maximum)偏低,S平均值(mean S-score) 小于0.5,位于0.1~0.2之间,表明该蛋白不含信号肽(图5),不是分泌型蛋白,推测该蛋白在细胞内不进行跨膜运输。膜蛋白分为胞外区、跨膜部分及胞内区,跨膜区就是蛋白在细胞膜内的部分。利用TMpred和SOSUI 分析跨膜性,结果表明该蛋白不存在跨膜蛋白,是一种可溶性蛋白(图6)。进一步利用PSORT软件对TfdA蛋白的细胞内定位进行预测,结果见表2,显示其定位在细胞质的可能性极大。
疏水作用是蛋白质折叠的主要驱动力,可为蛋白质二级结构预测提供佐证。利用ProtScale分析蛋白质的亲疏水性,287个氨基酸组成的TfdA蛋白中,亲水氨基酸占63.4%,而疏水氨基酸占33.8%,亲水性最强的氨基酸为第77位的丝氨酸,其分值最低(-2.311);疏水性最强的氨基酸为第130位的丝氨酸, 其分值最高(1.911);其亲水性区域较多,预测TfdA蛋白为亲水性蛋白(图 7)。这与缺乏疏水性的跨膜结构域 、 不具跨膜功能的预测相一致 。 因而C.campinensis BJ71菌株的TfdA蛋白是一个不含信号肽、定位于细胞质中的可溶性亲水蛋白。
.png) 图 5 SignalP 4.1 预测C. campinensis BJ71 菌株TfdA蛋白的信号肽 |
.png){kind=link}
.png) 图 6 C. campinensis BJ71 菌株 TfdA 蛋白跨膜性分析 |
.png) 图 7 ProtScale预测C. campinensis BJ71菌株TfdA蛋白的亲疏水性 |
.png) 表 2 C. campinensis BJ71 菌株 TfdA 蛋白的亚细胞定位分析 |
1.4 TfdA蛋白二级结构及模体分析
以C. necator JMP134菌株的TfdA蛋白作为参照,利用SOPMA分析2个不同菌株TfdA蛋白的二级结构。结果表明(表 3),2个菌株构成的二级结构元件中,α-螺旋(H)和 β-转角(T)比例存在微小差异。我们克隆获得的 BJ71菌株TfdA蛋白 α-螺旋为91个, β-转角为8个,无规卷曲(C)为136 个,延伸链(E)为52个;α-螺旋和无规卷曲是TfdA蛋白整体结构中的主要组成结构元件(图8),有利于稳定蛋白质的结构。
运用PredictProtein预测该蛋白是否有二硫键(Ceroni et al., 2006),结果显示,该蛋白没有二硫键形成 ;Prosite进行功能位点的分析(Bairoch et al., 1997),发现有1个糖基化位点 、6 个磷酸化位点、2个酰基化位点和2个酰胺化位点(表 4)。蛋白质翻译后修饰在酶和其它的重要功能分子活性的发挥、 第二信使传递和酶的级联作用中起到重要的作用,其中,蛋白质磷酸化是蛋白质翻译后修饰的重要内容,显然,该蛋白磷酸化位点数明显高于糖基化位点。
.png) 图 8 SOMPA分析C. campinensis BJ71 菌株TfdA蛋白的二级结构 |
.png) 表 3 2个菌株 TfdA 蛋白二级结构预测结果 |
.png) 表 4 C. campinensis BJ71菌株TfdA蛋白模体分析 |
1.5 TfdA蛋白卷曲螺旋域分析
卷曲螺旋是由2~5条α螺旋相互缠绕而形成的平行或反平行超螺旋结构,含有卷曲螺旋结构的蛋白主要是一些转录因子、结构蛋白、膜蛋白和酶等(魏香等, 2004)。亮氨酸拉链即为典型的卷曲结构。虽然α螺旋在该蛋白中的比例较大(31.71%),但COILS预测结果显示并不含卷曲螺旋域,也不形成亮氨酸拉链(图 9)。
.png) 图 9 COILS分析C. campinensis BJ71菌株TfdA蛋白的卷曲螺旋域 |
1.6 TfdA蛋白三级结构预测及分析
利用RaptorX软件进行三级结构预测,结果显示可以PDB数据库中3pvjA、1gqwA和1oihA为模板,最优模板为3pvjA,将具有27个氨基酸差异的 BJ71菌株TfdA蛋白以及 C. necator JMP134 TfdA蛋白(NCBI登录号:YP_025400)同时建模,三级结构如图10。可以看出,2 个蛋白的三级结构极其相似,但JMP134菌株的TfdA蛋白似乎具有更多的β-转角(图中箭头所示);前面利用SOPMA预测 JMP134菌株TfdA蛋白二级结构的结果也表明α-螺旋、β-转角比例分别略低于和高于BJ71菌株的TfdA蛋白,这也与预测的三级结构吻合。使用MolProbity软件中Ramachandran分析BJ71菌株TfdA的三级结构(图11),显示构建的TfdA三级结构模型有92.3% (263/285)的氨基酸位点都位于最佳区域内,有97.9% (279/285)的氨基酸位点位于允许区域范围内,说明本次模型结构可靠准确。
.png) 图 10 RaptorX预测TfdA蛋白三级结构 |
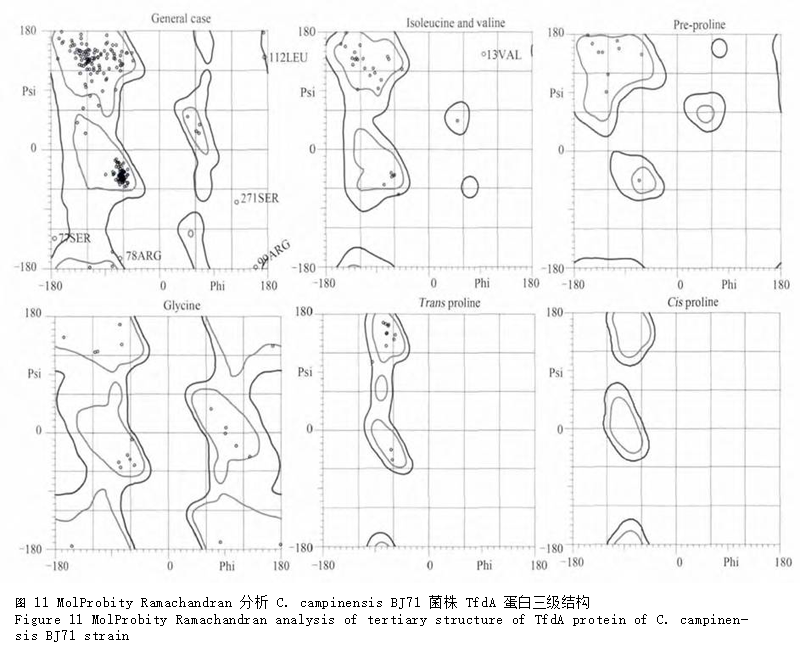 图 11 MolProbity Ramachandran分析C. campinensis BJ71 菌株TfdA蛋白三级结构 |
2 讨论
借助于生物信息学手段,我们对从2,4-D高效降解菌C. campinensis BJ71菌株中克隆获得的2,4-D降解基因tfdA及其编码产物进行了一系列分析,结果表 明该基因全长864 bp、属于Ⅰ族tfdA基因;其编码产物为包含287个氨基酸的Fe(Ⅱ)/α-酮戊二酸依赖性双加氧酶 TfdA蛋白,分子量为32 KD、理论等电点pI为6.01。该蛋白不含信号肽和跨膜结构域,是一种定位于细胞质中的可溶性亲水蛋白。α螺旋和无规卷曲是其二级结构的主要组成元件,内部存在一系列磷酸化位点和较少的糖基化、酰基化和酰胺化位点。
蛋白质三级结构预测的方法通常有同源建模法、折叠识别法和从头预测法。同源建模法(homology modeling)是基于待建模蛋白在模板库中可找到序列较相似的蛋白而进行预测的,又称为比较建模(comparative modeling),其代表软件为SWISS-MODEL,有研究显示如果序列相似性高于75%则预测准确度较高,低于40%则需要手动调整(Schwede et al., 2003; Arnold et al., 2006)。从头计算方法(ab initio)的理论依据是依据热力学理论,即求蛋白质能量最小的状态,但该方法并不实用,目前仅能计算低于120个氨基酸形成的结构。针对较低序列相似性的折叠识别法(fold recognition),又称为穿线法(threading method), 以加拿大滑铁卢大学的RAPTOR为代表;该方法在CASP (critical assessment of protein structure prediction)评估中被评价为最好的结构预测软件,可以有效优化蛋白穿线打分功能(Xu and Li, 2003)。与老版本的RAPTOR程序相比,RaptorX有3个提高,第一是考虑了蛋白特征之间的相关性;第二是通过借力结构信息处理相似性不高的蛋白;第三是利用了新的多重模板打分运算以扩展模板结构相似性和提高比 对准确性。这种基于多个模板进行折叠识别的特征, 大大提高了预测结构的准确性(Peng and Xu, 2009; 2010; 2011; Kllberg et al., 2012)。
本研究中,我们试图利用SWISS-MODEL进行同源建模,但发现与PDB数据库中的模板同源性仅有30.822%,因而并不适合采用该方法建模,而Rap-torX适合于同源性较低的蛋白序列三级结构的预测,应用该方法进行蛋白三维结构预测、经Ramachandran分析获得的结构合理可靠。Knauer等(2012)的研究表明,P. putida KT2440与E. coli的TauD晶体结构(PDB ID: 3pvjA, 1gqw)均为四聚体。 以 PDB 数据库中3pvjA为模板,利用RaptorX进行建模,发现C. campinensis BJ71菌株TfdA蛋白与C. necator JMP134菌株的三级结构非常相似,但 α-螺 旋和β-转角的比例略有不同,可能会造成2个TfdA蛋白在2,4-D降解能力上存在一定的差异。本研究可为该蛋白的原核表达、设计突变位点进行突变研究奠定基础。
3 材料与方法
3.1 材料
分析用的2,4-D降解基因tfdA (GenBank登录号: KJ028765)核苷酸序列是从本实验室前期分离筛选到的2,4-D高效降解菌Cupriavidus campinensis BJ71菌株(保藏号: CCTCC M 2014006)中克隆得到的。
3.2 tfdA 基因编码产物保守域及系统发生分析
利用Bioedit将克隆获得的tfdA基因核苷酸序列翻译为蛋白质序列;利用NCBI网站上CD Search(http://www.ncbi.nlm.nih.gov/Structure/cdd/wrpsb.cgi)分析编码产物保守结构域;选择Alacligenes eutrophus tfdA (M16730)、Burkholderia sp. RASC tfdA (U25717)、 Burkholderia cepacia 2a tfdA (AF029344)分别为Class Ⅰ、Ⅱ、Ⅲ tfdA基因的代表,Alphaproteobacterium sp. HW13 tfdAα (AB074492)为tfdA-like基因的代表,以Escherichia coil str. K-12 substr. MG1665 tauD (NC_ 000913)为外群;利用ClustalW 2.0 (Larkin et al., 2007)进行核苷酸多重序列比对,用MEGA5.0软件采用邻近法(Neighbor-Joining)进行系统发育分析、构建系统发育树(Tamura et al., 2011),通过重复取样1 000次进行自展值(Bootstrap)分析来评估系统发育树的拓扑结构稳定性。
3.3 tfdA基因编码产物的基本理化性质分析
在ExPASy网站上利用在线软件Protparam(http://www.expasy.org/tools/protparam.html)分析tfdA基因编码产物的基本理化性质(Gasteiger et al., 2005)。
3.4 tfdA基因编码产物信号肽、跨 膜区、定位及亲疏水性分析
利用SignalP 4.1 Server (http://www.cbs.dtu.dk/services/SignalP) (Petersen et al., 2011)预测 tfdA基因编码产物有无信号肽序列;利用TMpred (http://www.ch.embnet.org/software/TMPRED_form.html) (Hofmann and Stoffel, 1993)分析蛋白跨膜区、SOSUI (http://bp.nuap.nagoya-u.ac.jp/sosui/) (Hirokawa et al., 1998)分析是否为可溶蛋白;利用PSORT (http://psort.hgc.jp)进行蛋白定位分析; 利用ExPASy网站上ProtScale (http://ca.expasy.org/tools/protscale.html) (Gasteiger et al., 2005)分析亲疏水性。
3.5 tfdA基因编码产物二级结构及模体分析
蛋白质二级结构是指多肽链有规则重复的构象,其结构组件为α螺旋、β转角、无规卷曲和延伸链。利用PBILIBCP LyonGerland网站上SOMPA (http://npsa-pbil.ibcp.fr/cgi-bin/npsa_automat.pl?page=npsa_sopma.html) (Geourjon and Deléage, 1995) 进行二级结构预测;利用PredictProtein (http://www.predictprotein.org/) (Rost et al., 2004)进行功能预测、二硫键及结构域;利用COILS (http://www.ch.embnet.org/software/COILS_form.html) (Lupas et al., 1991) 分析蛋白有无卷曲螺旋域结构。
3.6 tfdA基因编码产物三级结构模拟
利用在线软件RaptorX(http://raptorx.uchicago.edu) (Kllberg et al., 2012)进行三级结构预测;用MolProbity软件 (http://molprobity.biochem.duke.edu; http://kinemage.biochem.duke.edu) (Davis et al., 2007; png et al., 2010)评估建模模型。
作者贡献
韩丽珍负责基因克隆、分析与论文撰写,李翠翠 负责资料收集并参与分析,责任作者赵德刚负责课 题整体设计、论文修改和指导。
致谢
本研究由国家转基因生物新品种培育重大专项子课题《安全转基因技术研究-转基因农作物的“基因删除”技术和“基因拆分”技术》(2013ZX08010003)、国家转基因生物新品种培育重大专项《安全转基因技术研究》子项目《保障转基因生物安全的外源基因清除技术研究》(2014ZX0801008B-002)和贵州省科技厅基金项目《除草剂2,4-D降解基因tfdA的克隆及降解机制研究》(黔科合 J 字[2013]2123)共同资助。
Arnold K., Bordoli L., Kopp J., and Schwede T., 2006, The SWISS-MODEL workspace: A web-based environment for protein structure homology modelling, Bioinformatics, 22 (2): 195-201
Chen V.B., Arendall Ⅲ W.B., Headd J.J., Keedy D.A., Immormino R.M., Kapral G.J., Murray L.W., Richardson J.S., and Richardson D.C., 2010, Molprobity: All-atom structure validation for macromolecular crystallography, Acta Crystallogr D Biol. Crystallogr, D66: 12-21
Cox L., Celis R., Hermosín M.C., and Cornejo J., 2000, Natural soil colloids to retard simazine and 2,4-D leaching in soil, J. Agric. Food Chem., 48(1): 93-99
Davis I.W., Leaver-Fay A., Chen V.B., Block J.N., Kapral G.J., Wang X., Murray L.W., Arendall Ⅲ W.B., Snoeyink J., Richardson J.S., and Richardson D.C., 2007, MolProbity:All-atom contacts and structure validation for proteins and nucleic acids, Nucleic Acids Res., 35: W375-W383
Elkins J.M., Ryle M.J., Clifton I.J., Hotopp J.C.D., Lloyd J.S., Burzlaff N.I., Baldwin J.E., Hausinger R.P., and Roach P.L., 2002, X-ray crystral structure of Escherichia coli Taurine/α-ketoglutarate dioxygenase complexed to ferrous iron and substrates, Biochemistry, 41(16): 5185-5192
Farenhorst A., Londry K.L., Nahar N., and Gaultier J., 2008, In-field variation in 2,4-D mineralization in relation to sorption and soil microbial communities, J. Environ. Sci. Health B, 43: 113-119
Fukumori F., and Hausinger R.P., 1993, Alcaligenes eutrophus JMP134 “2,4-dichlorophenoxyacetate monooxygenase”is an α-ketoglutarate-dependent dioxygenase, J. Bacteriol., 175: 2083-2086
Gasteiger E., Hoogland C., Gattiker A., Duvaud S., Wilkins M.R., Appel R.D., and Bairoch A., 2005, Protein identification and analysis tools on the ExPASy Server, In: Walker J.M. (ed.), The proteomics protocols handbook, Humana Press, pp. 571-607
Geourjon C., and Deléage G., 1995, SOPMA: Significant improvements in protein secondary structure prediction by consensus prediction from multiple alignments, Comput. Appl. Biosci., 11(6): 681-684
Han L.Z., and Zhao D.G., 2012, Research progress of microbial degradation of the herbicide 2,4-dichlorophenoxyacetic acid, Nongyao (Agrochemicals), 51 (10): 710-714, 719 ( 韩丽珍, 赵德刚, 2012, 除草剂2,4-滴微生物降解研究进展, 农药, 51(10): 710-714, 719)
Hirokawa T., Boon-Chieng S., and Mitaku S., 1998, SOSUI: Classfication and secondary structure prediction system for membrane proteins, Bioinformatics, 14(4): 378-379
Hofmann K., and Stoffel W., 1993, TMbase-A database of mem brane spanning proteins segments , Biol . Chem . Hoppe-Seyler, 374: 166
Hogan D.A., Smith S.R., Saari E.A., McCracken J., and Hausinger R.P., 2000, Site-directed mutagenesis of 2,4-dichlorophenoxyacetic acid/α-ketoglutarate dioxygenase, J. Biol. Chem., 275(17): 12400-12409
Itoh K., Tashiro Y., Uobe K., Kamagata Y., Suyama K., and Ya- mamoto H., 2004, Root nodule Bradyrhizobium spp. harbor tfdA α and cadA, homologous with genes encoding 2,4-dichlorophenoxyacetic acid-degrading proteins, Appl. Environ. Microbiol., 70(4): 2110-2118
Johannesen H., and Aamand J., 2003, Mineralization of aged a- trazine, terbuthylazine, 2,4-D, and mecoprop in soil and aquifer, Environ. Toxicol. Chem., 22: 722-729
Kllberg M., Wang H., Wang S., Peng J., Wang Z., Lu H., and Xu J., 2012, Template-based protein structure modeling us- ing the RaptorX web server, Nat. Protoc., 7(8): 1511-1522
Kamagata Y., Fulthorpe R.R., Tamura K., Takami H., Forney L. J., and Tiedje J.M., 1997, Prisine environments harbor a new group of oligotrophic 2,4-dichlorophenoxyacetic acid-degrading bacteria, Appl. Environ. Microbiol., 63 (6): 2266-2272
Kitagawa W., Takami S., Miyauchi K., Masai E., Kamagata Y., Tiedje J.M., and Fukuda M., 2002, Novel 2,4-dichlorophe- noxyacetic acid degradation genes from oligotrophic Bradyrhizobium sp. strain HW13 isolated from a pristine en- vironment, J. Bacteriol., 184(2): 509-518
Knauer S.H., Hartl-Spiegelhauer O., Schwarzinger S., Hnzelmann P., and Dobbek H., 2012, The Fe ( Ⅱ )/α-ketoglu- tarate-dependent taurine dioxygenases from Pseudomonas putida and Escherichia coli are tetramers, FEBS J., 279: 816-831
Larkin M.A., Blackshields G., Brown N.P., Chenna R., McGettigan P.A., McWilliam H., Valentin F., Wallace I.M., Wilm A., Lopez R., Thompson J.D., Gibson T.J., and Higgins D.G., 2007, Clustal W and Clustal X version 2.0, Bioinformat- ics, 23(21): 2947-2948
Lupas a., van Dyke M., and Stock J., 1991, Predicting coiled coils from protein sequences, Science, 252(5009): 1162-1164 McGowan C., Fulthorpe R., Wright A., and Tiedje J.M., 1998, Evidence for interspecies gene transfer in the evolution of 2,4-dichlorophenoxyacetic acid degraders, Appl. Environ. Microbiol., 64(10): 4089-4092
Müller T.A., Fleischmann T., van der Meer J.R., and Kohler H.P. E., 2006, Purification and characterization of two enantiose- lective α-ketoglutarate-dependent dioxygenases, RdpA and SdpA, from Sphingomonas herbicidovorans MH, Appl. Environ. Microbiol., 72(7): 4853-4861
Peng J., and Xu J., 2009, Boosting protein threading accuracy, Res. Comput. Mol. Biol., 5541: 31-45
Peng J., and Xu J., 2010, Low-homology protein threading, Bioinformatics, 26(12): i294-i300
Peng J., and Xu J., 2011, Raptor X: Exploiting structure informa- tion for protein alignment by statistical inference, Proteins, 79(Suppl 10): 161-171
Petersen T.N., Brunak S., von Heijne G., and Nielsen H., 2011, SignalP 4.0: Discriminating signal peptides from transmem-brane regions, Nat. Methods, 8: 785-786
Rost B., Yachdav G., and Liu J., 2004, The PredictProtein server, Nucleic Acids Res., 32: W321-W326
Schwede T., Kopp J., Guex N., and Peitsch M.C., 2003, SWISS-MODEL: An automated protein homology-modeling server, Nucleic Acids Res., 31(13): 3381-3385
Starrett S.K., Christians N.E., and Austin T.Al., 2000, Movement of herbicides under two irrigation regimes applied to turf- grass, Adv. Environ. Res., 4(2): 169-176
Tamura K., Peterson D., Peterson N., Stecher G., Nei M., and K umar S., 2011, MEGA5: Molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods, Mol. Biol. Evol., 28(10): 2731-2739
Wei X., Zeng X.G., and Zhou H.M., 2004, Progress on the study of coiled coils in protein structures, Zhongguo Shengwu Huaxue Yu Fenzi Shengwu Xuebao (Chinese Journal of Biochemistry and Molecular Biology), 20(5): 565-571 (魏香, 曾宪 纲, 周海梦, 2004, 蛋白质结构中卷曲螺旋的研究进展, 中国生物化学与分子生物学报, 20(5): 565-571)
Xu J., and Li M., 2003, Assessment of RAPTOR's linear pro- gramming approach in CAFASP3, Proteins, 53: 579-584
Zaprasis A., Liu Y.J., Liu S.J., Drake H.L., and Horn M.A., 2010, Abundance of novel and diverse tfdA-like genes, encoding putative phenoxyalkanoic acid herbicide-degrading dioxyge- nases, in soil, Appl. Environ. Microbiol., 76(1): 119-128