SSR数据处理宏程序DataTrans 1.0 
2.中国农业科学院烟草研究所/中国农业科学院烟草遗传改良与生物技术重点开放实验室, 青岛, 266101
 作者
作者  通讯作者
通讯作者
《分子植物育种》网络版, 2011 年, 第 9 卷, 第 48 篇 doi: 10.5376/mpb.cn.2011.09.0048
收稿日期: 2011年03月10日 接受日期: 2011年04月13日 发表日期: 2011年04月21日
盖红梅等, 2011, SSR数据处理宏程序DataTrans 1.0, 分子植物育种 Vol.9 No.48 (doi: 10.5376/mpb.cn.2011.09.0048)
随着群体遗传学研究的不断深入和高通量基因分型技术的成功实施,越来越多的研究需要处理大量的SSR数据。而原始数据格式往往与不同分析软件的输入格式之间存在较大差别,由此,SSR数据前期处理日益成为数据分析过程中的限速步骤。为此,本研究应用Excel的VBA语言开发了一套进行SSR数据处理的宏程序DataTrans1.0。该程序可以实现由原始的bp数据,转换为Ntsys软件需要的0, 1格式、Popgene软件需要的基因型格式及PowerMarker、Structure和Tassel等软件的输入格式;还可以实现由0, 1数据转换为Popgene软件需要的基因型格式。与人工转换相比,DataTrans 1.0大大提高了数据转换的效率和准确性,是广大群体遗传学和基因组学研究人员不可或缺的重要桥梁工具。
微卫星DNA又叫简单重复序列(Simple Sequence Repeats, SSR),具有数量丰富,覆盖整个基因组,多态性高且呈共显性遗传等优点,因此广泛地应用于遗传多样性分析(Vigoroux et al., 2005; 盖红梅等, 2005; 任民等, 2005)、遗传图谱构建(Röder et al., 1998; Somers et al., 2004)、核心种质构建 (Hao et al., 2008)、QTL定位(Yang et al., 2007; Su et al., 2006; Hanocq et al., 2007)、关联分析(Remington et al., 2001; Breseghello and Sorrells, 2006; Zhang et al., 2007a, b; 王兰芬等, 2007)、育种亲本评估(盖红梅等, 2009)等研究中。随着分子生物学的不断发展,SSR数据的产生也进入高通量水平。与此同时,各种群体遗传学分析软件(包)应运而生,比如Ntsys (Rohlf, 2002) (http://www.exetersoftware.com/cat/nysyspc.html),Popgene (Ye et al., 1999) (http://www.ualberta.ca/~fyeh/index.htm),PowerMarker (Liu and Muse, 2005) (http://www.PowerMarker.net/), Structure (Pritchard, 2000) (http://pritch.bsd.uchicago.edu/)及Tassel (Buckler, 2007) (http://www.maizegenetics.net/bioinformatics/tassel/)等。但是不同的软件,其数据的输入格式不同,而没有正确的输入格式就无法运行相应的软件,从而无法对数据进行深入剖析。因此,如何将SSR原始采集数据转换为不同遗传学软件所需的数据格式成为一个繁琐、枯燥、但又不可缺少的步骤,这让众多学者感到非常棘手。虽然,应用Excel进行人工转换或利用其查找、替换和函数也能实现数据格式转换功能,但是转换效率非常低,而且容易出错。因此,在基因型扫描的高通量时代,面临越来越多的微卫星数据,急需开发一款能够快速、高效、准确的将SSR原始保存数据转换为遗传分析软件所需格式的软件。目前,已有一些群体遗传学软件带有数据转换的功能,如Genetix (Belkhir et al., 2001),MSTools3 (http://animalgenomics.ucd.ie/sdepark/ms-toolkit/)等,但Genetix的格式转换并非原始数据转换而是等位变异频率数据的转换,且只有法语版本,无法满足广大中国学者的需要。而MSTools3针对Arlequin,GenePop,Microsat,Fstat和Dispan等软件的需要设计的,随着分析软件的更新换代,现有软件已不能满足多数用户的需求。
Excel是微软公司的办公软件Microsoft office的一个重要组件,它可以进行各种数据的处理、统计分析,广泛应用于多个领域,多数用户的SSR原始数据就保存于Excel。因此,对Excel进行二次开发,能给用户带来很大方便。Excel内嵌的Microsoft VBA (Visual Basic for Application)语言是以Visual Basic为基础的编程语言,直观、易用、能与Excel强大的电子表格和函数功能无缝结合。同时对计算机软硬件的要求也比较低,占用系统资源少,可开发绿色软件,因此,该语言在多个领域得到了广泛应用。刘仁虎和孟金陵(2003)采用VBA语言实现了在Excel中进行遗传连锁图的绘制;Kemmer和Keller (2010)用该语言实现了非线性最小二乘数据的拟合。
因此,本研究在明确目前常用的5款群体遗传学分析软件(Ntsys,Popgene,PowerMarker,Structure和Tassel)的SSR数据输入格式的基础上,使用Microsoft VBA语言,开发了SSR数据处理程序DataTrans 1.0。该软件简化了SSR原始数据到这5款群体遗传学分析软件的格式转换,大大节省了数据分析时间,提高了数据转换的准确性,同时软件界面友好,简单易用,对SSR数据的深入挖掘分析提供了有力保障。用户可以通过以下网址获取该程序:http://u.115.com/file/f2ebd280f。
1需求分析
1.1 SSR数据来源及其原始格式
目前广泛采用的SSR检测方法为聚丙烯酰胺凝胶电泳(图1)和毛细管凝胶电泳(图2)。其中前者一般按扩增带的有无采集为0,1格式的数据(表1),也可以根据分子量内标而获得bp(碱基数)格式的数据;毛细管电泳的原始数据通常以bp格式保存(表2),缺失数据记为9。
图1 聚丙烯酰胺凝胶电泳扩增片段, 有带记为1无带记为0 Figure 1 Bands pattern from 6% PAGE, “1” for band presence and “0” for band absence |
图2 毛细管凝胶电泳扩增片段峰图(橘黄色峰为分子量内标) Figure 2 The sequence trace files from ABI 3730 (The orange peak indicate molecular weight of internal standard) |
 表1 SSR数据0, 1格式示例 Table 1 The SSR data of “0, 1” type |
.png) 表2 SSR数据bp格式示例 Table 2 The SSR data of size (bp) type |
1.2常用群体遗传学分析软件的输入格式
Ntsys通常处理0,1格式的数据;Popgene的输入文件通常以基因型数据为基础,例如AA代表一个纯合位点,AB代表一个杂合位点;PowerMarker,Structure和Tassel等软件的数据输入格式都基于bp值。以下为上述软件的输入格式示例。
1.2.1 Ntsys的输入文件格式
1 4b 11 1 9
Line1 1 0 0 0 1 0 1 0 1 0 0
Line2 1 0 1 0 9 9 9 9 0 1 0
Line3 0 1 0 0 0 1 0 0 1 0 0
Line4 0 0 0 1 0 0 0 1 0 0 1
1.2.2 Popgene输入文件格式
/*Input your note here*/
Number of populations=1
Number of loci=3
Locus name: marker1 marker2 marker3
AA AC AA
AC 99 BB
BB BB AA
DD DD CC
1.2.3 PowerMarker输入文件格式
Code Marker1 Marker2 Marker3
Line1 190/190 200/210 236/236
Line2 190/194 9/9 238/238
Line3 192/192 208/208 236/236
Line4 196/196 212/212 240/240
1.2.4 Structure输入文件格式
Marker1 Marker2 Marker3
Line1 190 200 236
Line1 190 210 236
Line2 190 9 238
Line2 194 9 238
Line3 192 208 236
Line3 192 208 236
Line4 196 212 240
Line4 196 212 240
1.2.5 Tassel输入文件格式
4 3:2
Marker1 Marker2 Marker3
Line1 190:190 200:210 236:236
Line2 190:194 ?:? 238:238
Line3 192:192 208:208 236:236
Line4 196:196 212:212 240:240
1.3设计目标
应处理从SSR原始数据保存格式转换为Ntsys,Popgene,PowerMarker,Structure和Tassel软件所需的数据格式,而且要求准确无误、快速高效;另外,需有自己的操作界面,通过点击命令按钮进行操作,简单易用;最后,对用户友好,无需安装,不在系统文件夹添加文件、占用系统资源小。
2 DataTrans1.0简介
DataTrans1.0是一款SSR数据前期处理的Excel宏程序,可以将SSR原始采集数据准确、高效的转换为多款统计分析软件(Ntsys, Popgene, PowerMarker, Structure和Tassel)所需的数据格式。该程序以VBA为开发语言,在Excel环境下运行,界面友好(图3),操作简便,是群体遗传学和基因组学数据分析的重要桥梁工具。与手工转换和使用Excel函数相比,使用该软件对提高工作效率和确保数据的准确性方面具有极大的优势(表3)。目前多数学者采用的是Excel 2003,但是由于其仅能容纳256列数据,已经无法满足高通量的SSR数据保存。而Excel 2007能容纳1048576行和16384列的数据,更适合高通量数据的存储。因此,DataTrans1.0开发了DataTrans1.0(2003)和DataTrans1.0(2007)两个版本,以满足不同用户的需求。这两个版本在软件的功能、界面和使用方法上完全相同,两者的区别仅在于DataTrans1.0 (2007)能处理更多的SSR数据,可充分满足高通量数据分析的要求。本文将以DataTrans1.0 (2007)为例进行介绍。
.png) 图3 DataTrans1.0(2007)的用户界面 Figure 3 The user interface of DataTrans1.0 (2007) |
 表3 DataTrans1.0能够大大提高SSR数据前期处理的效率 注: DataTrans 1.0的结果为实际运算, Excel函数和Excel查找、替换的结果为根据经验估计 Table 3 Data Trans1.0 can raise the efficiency of pre-processing of SSR data greatly Note: The data point of DataTrans1.0 obtained by real operation, the data point of Excel function and searching/shifting based on empirical estimation |
3 DataTrans1.0的使用方法
3.1开启Excel宏
在运行该程序前应确保Excel已经开启宏,否则需要手动开启。方法为:在Excel 2003中,进入“工具/宏/安全性”将其设置为“低”;在Excel 2007中,进入“Excel选项/信任中心/信任中心设置/宏设置”选择“启用所有宏”。
3.2 bp型原始数据输入格式
按照多数用户的SSR原始数据保存习惯,将引物名称放在第一行,材料编号放在第一列,材料n在引物m的bp值则放在n和m的交界处,由于输入的为二倍体数据,因此引物以空白间隔排列,将整理好的原始数据放在sheet1(图4)。
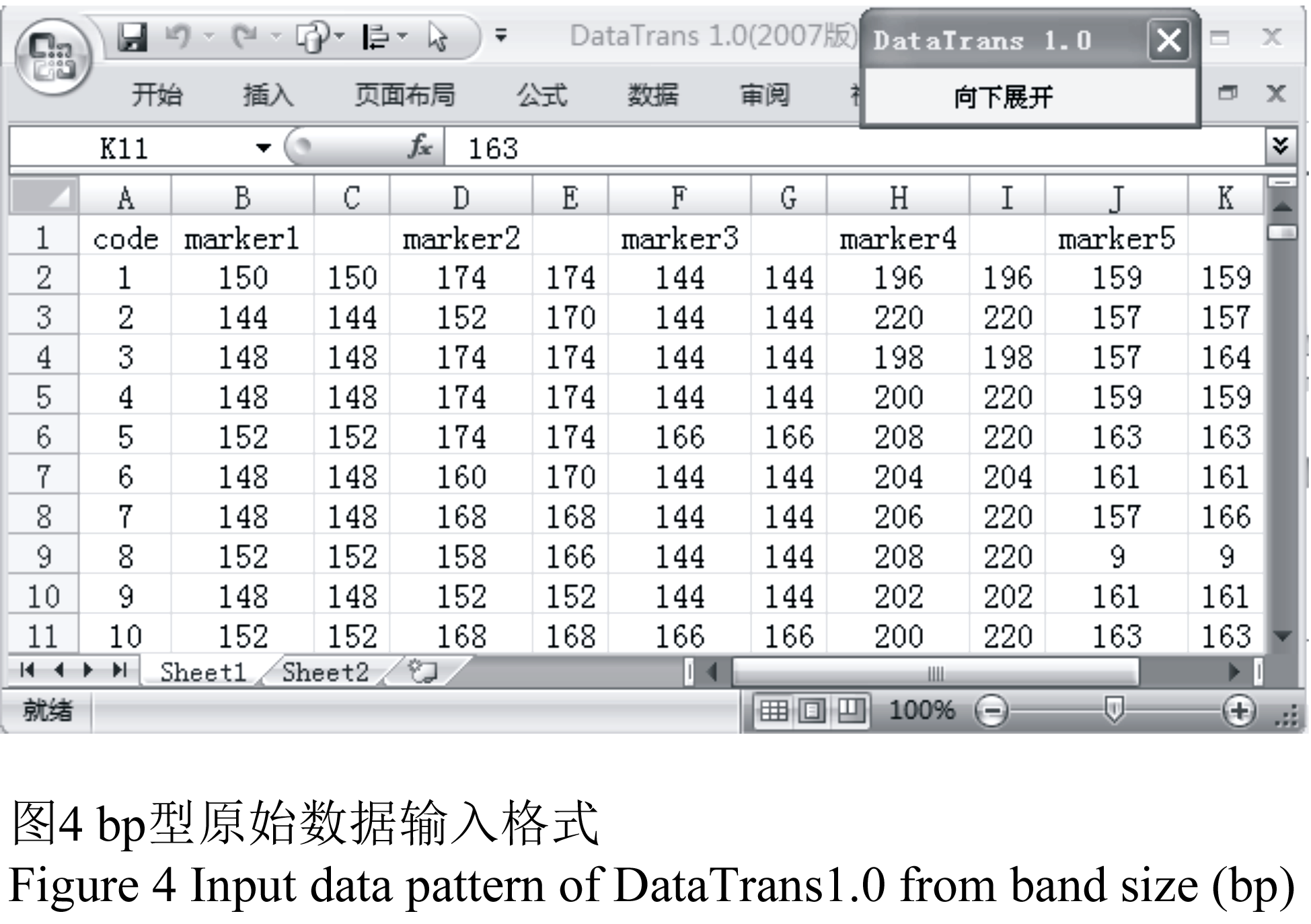 图4 bp型原始数据输入格式 Figure 4 Input data pattern of DataTrans1.0 from band size (bp) |
3.3 0,1型原始数据输入格式
以0, 1记载的SSR原始数据,需将引物名称放在第一行,每个引物的扩增条带数放在第二行,与相应的标记对齐,材料编号放在第一列。材料n在引物m的第j条带处有带标为1,无带标为0,缺失标为9,将整理好的原始数据放在sheet1(图5)。
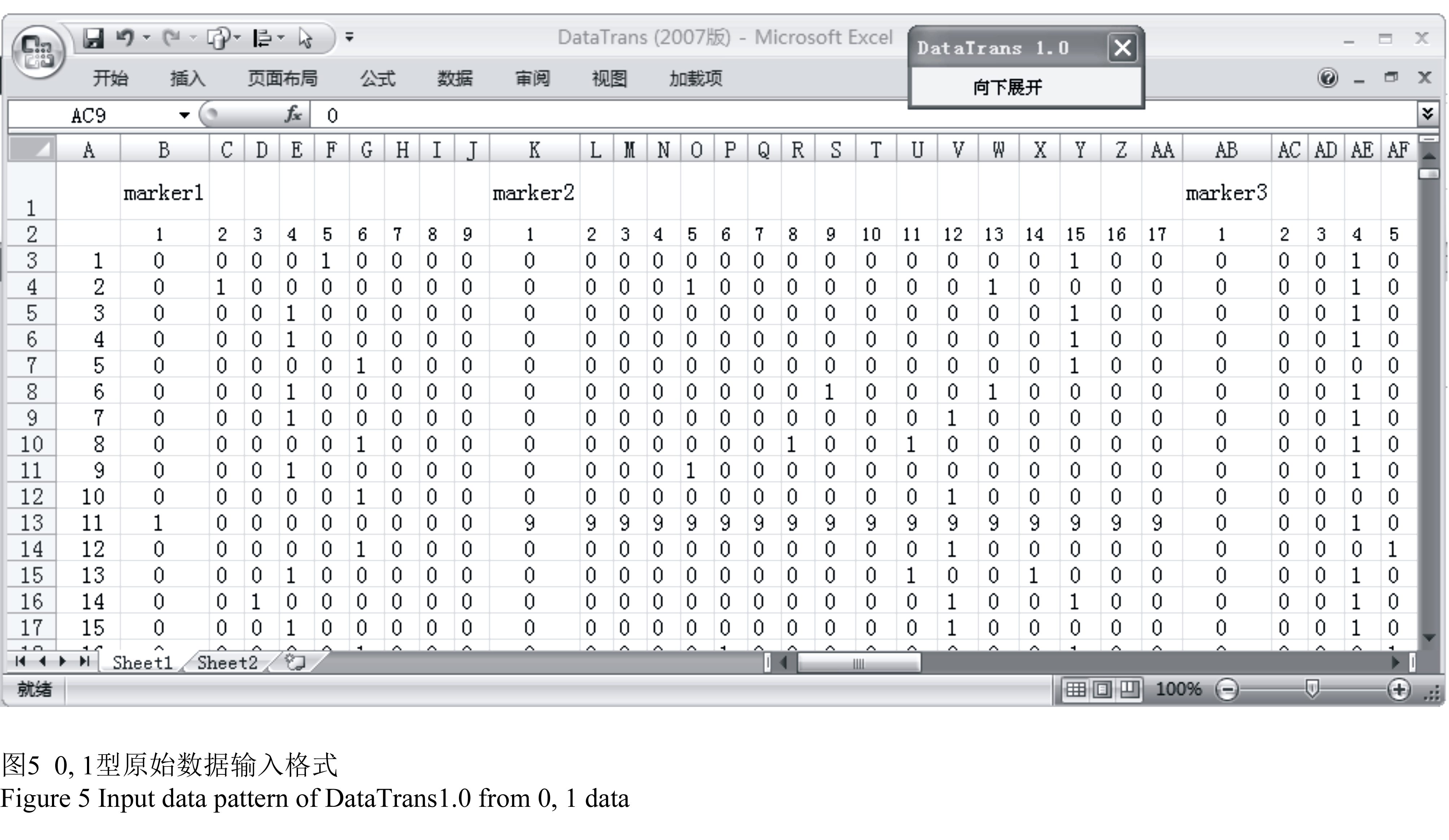 图5 0, 1型原始数据输入格式 Figure 5 Input data pattern of DataTrans1.0 from 0, 1 data |
3.4不同SSR数据类型间的转换
打开DataTrans1.0软件,将需要转换的原始数据放入sheet1(图4; 图5)。如果原始保存数据为bp型(图4),则单击用户交互窗口的“bp型数据”选项卡,点击相应的命令按钮即可进行格式转换。比如:需要生成Structure软件所需数据,则单击“bp型数据”选项卡下的“转换成Structure格式”命令按钮,转换后的数据将自动保存在sheet 2中。如果原始保存数据为0, 1型(图5),则单击用户交互窗口的“0, 1型数据”选项卡,单击“转换成基因型数据”命令按钮,则Popgene软件所需的数据格式便在sheet 2中生成(图6)。
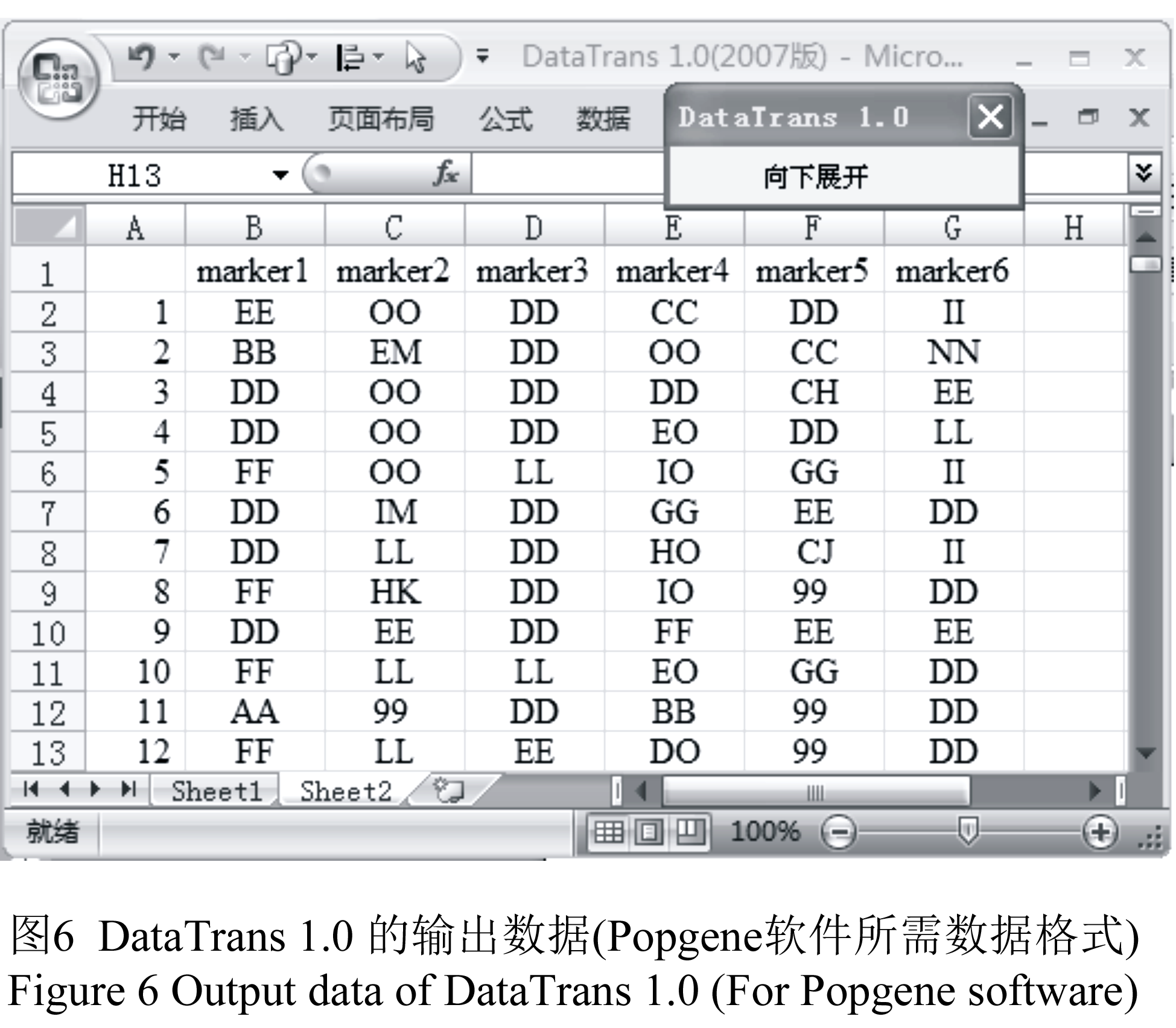 图6 DataTrans 1.0 的输出数据(Popgene软件所需数据格式) Figure 6 Output data of DataTrans 1.0 (For Popgene software) |
3.5数据后期处理
利用DataTrans 1.0实现了在Excel中进行SSR数据主体的格式转换,其生成数据存储在sheet 2中。而后仅需将sheet2中的数据拷贝到文本文件编辑器(如记事本, Editplus等)中,加入相应的参数即可。
4讨论
微卫星标记由于数量丰富,覆盖整个基因组,多态性高且呈共显性遗传等优点而被广泛应用于动植物QTL定位、关联分析及群体遗传学研究。随着研究的深入,对微卫星标记检测位点数的要求越来越高,而高通量基因型检测的成功实施使检测大量微卫星标记成为可能。随之而来的是海量的SSR数据的整理总结及多层面分析,因此必然会用到多种群体遗传学软件和软件包,比如Ntsys、Popgene、PowerMarker、Structure、Tassel等。但不同的软件往往需要不同的输入格式,即SSR原始采集数据必须转换多种格式进行运算,这给数据分析增添很多负担。因此,DataTrans 1.0软件的开发解决了SSR数据格式人工转换费时、费力且容易出错的难题,为SSR数据的深入分析提供了有力保障。
DataTrans 1.0软件基于Excel的VBA语言,以Excel为载体,拥有简洁的操作界面,因此,只要用户能进行Excel的基本操作,便能直接利用DataTrans 1.0进行数据格式转换,节省了大量学习相应软件输入格式和数据整理的时间,同时极大的提高了转换的准确性。原始数据量越大,越能体现出Datatrans 1.0的方便、高效和准确的优势,应用该软件能在数分钟之内完成人工数月之久的工作。另外,基于Excel宏开发的Datatrans 1.0还具有无须安装、不产生系统垃圾、可随意拷贝、节省磁盘空间等对用户十分友好的特性,是广大群体遗传学研究人员不可或缺的桥梁工具。
与其他数据转换软件如Genetix,MSTools 3等相比较,DataTrans 1.0对用户更加友好,可以实现从原始的“bp”数据或“0, 1”数据开始转换,符合多数学者对SSR数据的保存和使用习惯,而Genetix的格式转换并非原始数据转换而是等位变异频率数据的转换,且为法语版本。此外在支持的文件格式上也有较大区别,DataTrans 1.0对新近的一些常用分析软件提供了更多的支持,有利于研究人员采用新软件、新算法进行数据分析。今后DataTrans 1.0还会进一步完善,将向着对更多分析软件提供支持和更加方便友好的方向发展。
作者贡献
盖红梅和任民是本研究的实验设计和实验研究的执行人,盖红梅完成论文初稿的写作,任民完成论文的修改。全体作者都阅读并同意最终的文本。
致谢
感谢青岛市科技计划(10-3-4-14-1-jch,09-1-1-69-nsh),中央级公益性科研院所基本科研业务费专项(0032010038)和国家小麦现代产业技术体系建设专项经费(2060302-2-2)资助。感谢中国农业科学院作物科学所张学勇研究员的示例数据支持和对文稿的修改润色,王兰芬副研究员在DataTrans 1.0开发过程中给予的宝贵建议和意见。
参考文献
Belkhir K.P., Borsa P., Chikhi L., Raufaste N., and Bonhomme F., 2004, GENETIX 4.05, logiciel sous Windows TM pour la genetique des populations, Laboratoire Genome, Populations, Interactions, CNRS UMR 5171, Universite de Montpellier II, Montpellier (in France)
Breseghello F., and Sorrells M.E., 2006, Association mapping of kernel size and milling quality in wheat (Triticum aestivum L.) cultivars, Genetics, 172: 1165-1177 doi:10.1534/genetics.105.044586 PMid:16079235 PMCid:1456215
Ge H.M, Chen C.B., Shen F.F., Zhang W.X., Ren M., Wang Y.W., and Yang Q.W., 2005, Genetic diversity and conservation strategy of Oryza rufipogon along the Haojiang river in Guangxi Zhuang Autonomous Region, Zhiwu Yichuan Ziyuan Xuebao (J. Plant Genet. Res.), 6(2): 156-162 (盖红梅, 陈成斌, 沈法富, 张万霞, 任民, 王玉薇, 杨庆文, 2005, 广西武宣濠江流域普通野生稻居群遗传多样性及保护研究, 植物遗传资源学报, 6(2): 156-162)
Ge H.M., Wang L.F., You G.X., Hao C.Y., Dong Y.C., and Zhang X.Y., 2009, Fundamental roles of cornerstone breeding lines in wheat reflected by SSR random scanning, Zhongguo Nongye Kexue (Sci. Agric. Sin.), 42(5): 1503-1511 (盖红梅, 王兰芬, 游光霞, 郝晨阳, 董玉琛, 张学勇, 2009, 基于SSR标记的小麦骨干亲本育种重要性研究, 中国农业科学, 42(5): 1503-1511)
Hanocq E., Laperche A., Jaminon O., Laine A.L., and Gouis J.L., 2007, Most significant genome regions involved in the control of earliness traits in bread wheat, as revealed by QTL meta-analysis, Theor. Appl. Genet., 114(3): 569-584 doi:10.1007/s00122-006-0459-z PMid:17171391
Hao C.Y., Dong Y.C., Wang L. F., You G.X., Zhang H.N., Ge H.M., Jia J.Z., and Zhang X.Y., 2008, Genetic diversity and construction of core collection in Chinese wheat genetic resources, Chinese Sci. Bull., 53(10): 1518-1526 doi:10.1007/s11434-008-0212-x
Kemmer G., and Keller S., 2010, Nonlinear least-squares data fitting in Excel spreadsheets, Nature Protocol, 5(2): 267-281 doi:10.1038/nprot.2009.182
PMid:20134427
Liu K., and Muse S.V., 2005, PowerMarker: integrated analysis environment for genetic marker data, Bioinformatics, 21(9): 2128-2129 doi:10.1093/bioinformatics/bti282 PMid:15705655
Liu R.H., and Meng J.L., 2003, MapDraw: a microsoft excel macro for drawing genetic linkage maps based on given genetic linkage data. (Hereditas), 2003, 25(3): 317-321 (刘仁虎和孟金陵, 2003, MapDraw, 在Excel中绘制遗传连锁图的宏, 遗传, 25(3): 317-321)
Pritchard J. K., Stephens M., and Donnelly P., 2000, Inference of population structure using multilocus genotype data, Genetics, 155: 945-959
PMid:10835412 PMCid:1461096
Remington D.L., Thornsberry J.M., Matsuoka Y., Wilson L.M., Whitt S.R., Doebley J., Kresovich S., Goodman M.M., and Buckler E.S., 2001,Structure of linkage disequilibrium and phenotypic associations in the maize genome, Proc. Natl. Acad. Sci. USA, 98(20): 11479-11484 doi:10.1073/pnas.201394398 PMid:11562485 PMCid:58755
Ren M., Chen C.B., Rong T.Z., Zhang W.X., Ge H.M., and Yang Q.W., 2005,Genetic Diversity of Oryza rufipogon Griff.in southeast region of guangxi in China, (J. Plant Genet. Res.), 6(1): 31-36 (任民, 陈成斌, 荣廷昭, 张万霞, 盖红梅, 杨庆文, 2005, 桂东南地区普通野生稻遗传多样性研究, 6(1): 31-36)
Röder M.S., Korzun V., Wendehake K., Plaschke J., Tixier M.H., Leroy P., Ganal M.W., 1998, A microsatellite map of wheat, Genetics, 49: 2007-2023
Rohlf F.J., 2002, NTSYS-pc. Numerical taxonomy and multivariate analysis system, Version 2.10. Exeter Software, New York
Somers D.J., Isaak P., and Edwards K., 2004, A high-density microsatellite consensus map for bread wheat (Triticum aestivum L.), Theor. Appl. Genet., 109: 1105-1114 doi:10.1007/s00122-004-1740-7 PMid:15490101
Su J.Y., Tong Y.P., Liu Q.Y., Li B., Jing R.L., Li J.Y., and Li Z.S., 2006, Mapping quantitative trait loci for post-anthesis dry matter accumulation in wheat, J. Integr. Plant Biol., 48(8): 938-944 doi:10.1111/j.1744-7909.2006.00252.x
Vigoroux Y., Mitchell S., Matsuoka Y., Hamblin M., Kresovich S., Smith J.S.C., Jaqueth J., Smith O.S., and Doebley J., 2005, An analysis of genetic diversity across the maize genome using microsatellites, Genetics, 160: 1617-1630
Wang L.F., Balfourier F, Hao C.Y., Exbrayat-vinson F., Dong Y.C., Ge H.M., and Zhang X.Y., 2007, Comparison of genetic diversity level between european and east-asian wheat collections using SSR markers, (Sci. Agric. Sin.), 40(12): 2667-2678 (王兰芬, Balfourier F, 郝晨阳, Exbrayat-vinson F., 董玉琛, 盖红梅, 张学勇, 2007, 欧洲与东亚小麦品种遗传多样性的比较分析, 中国农业科学, 40(12): 2667-2678)
Yang D.L., Jing R.L., Chang X.P., and Li W., 2007, Identification of Quantitative Trait loci and environmental Interactions for accumulation and remobilization of water-soluble carbohydrates in wheat (Triticum aestivum L.) stems, Genetics, 176: 571-584 doi:10.1534/genetics.106.068361 PMid:17287530 PMCid:1893045
Yeh F.C., Yang R.C., and Boyle T., Popgene version 1.31 quick user guide, Canada: University of Alberta, and Centre for Interna-tional Forestry Research, 1999
Zhang X.Y., Ge H.M., You G.X., Hao C.Y., Wang L.F., Dong Y.S., Tong Y.P., Li B., and Li Z.S., 2007b, Dissection of wheat breeding in China by marker/trait association, Molecular Plant Breeding, 5(2): 225-226
Zhang X.Y., Tong Y.P., You G.X., Hao C.Y., Ge H.M., Wang L.F., Dong Y.S., and Li Z.S., 2007a, Hitchhiking effect mapping: A new approach for discovering agronomic important genes, Sci. Agric. Sin., 6: 255-264