研究报告
扩增子测序结合高分辨率溶解曲线鉴定花生单核苷酸多态 
2国家油料作物改良中心南方花生分中心, 广州, 510640
3广东省农作物遗传改良重点实验室, 广州, 510640
4华南农业大学农学院, 广州, 510642
 作者
作者  通讯作者
通讯作者
豆科基因组学与遗传学, 2015 年, 第 6 卷, 第 6 篇
收稿日期: 2015年11月04日 接受日期: 2015年11月04日 发表日期: 2015年11月04日
引用格式(中文):
李杏瑜, 刘颖, 朱方何, 洪彦彬, 刘洪, 陈小平, 李海芬, 梁炫强, 2015, 扩增子测序结合高分辨率溶解曲线鉴定花生单核苷酸多态, 分子植物育种, 13(9): 1970-1979
引用格式(英文):
Li X.Y., Liu Y., Zhu F.H., Hong Y.B., Liu H., Chen X.P., Li H.F., and Liang X.Q., 2015, Identification and evaluation of Single-nucleotide polymorphisms in peanut (Arachis hypogaea L.) based on amplicon sequencing combined with high resolution melting analysis, Fenzi Zhiwu Yuzhong (Molecular Plant Breeding), 13(9): 1970-1979
异源四倍体花生(Arachis hypogaea L.)包含两套基因组,分别来自二倍体祖先A. duranensis (A基因组)和A. ipaensis (B基因组)。相对于二倍体,异源四倍体SNP的鉴定和分析面临更多挑战,因为在SNP的鉴定和分析过程中,通常需要同时分析两套基因组中相同位点的DNA序列。本研究以12个花生品种和2个二倍体祖先为材料,通过扩增子重测序EST和GSS (各100条序列)开发SNP。结果共检测出18个EST-SNPs和44个genomic-SNPs,出现频率分别为1 SNP/2577 bp和1 SNP/1011 bp。为了进一步评估和应用所开发的SNP,采用高分辨率溶解曲线方法对96个花生品种进行SNP基因分型。EST-SNP在供试品种中的多态性信息量介于0.021~0.413,平均为0.172。Genomic-SNP的多态性信息量介于0.08~0.478,平均为0.249。本研究表明采用扩增子测序和HRM方法能够从异源四倍体花生中准确鉴定SNP,且所开发的SNP信息量丰富,能够用于花生遗传育种研究。
单核苷酸多态(Single nucleotide polymorphisms, SNPs)是动植物基因组中最主要的多态类型,现已被大量用于遗传作图、基因定位和关联分析等研究。人类单倍体计划通过SNP进行标记-性状关联分析,为人类基因组中复杂性状的研究开辟新途径。近年来,一系列低成本、高通量的SNP基因分型平台正加快遗传学、基因组学等研究的发展。然而,当前SNP基因分型平台以及相应的数据分析软件主要针对人和二倍体生物进行设计,将其应用到植物上存在很多问题,因此自然界中多数植物为多倍体或古多倍体。
异源多倍体中,SNP被定义为不同个体同一亚基因组间的单核苷酸变异。部分同源序列变异(Homoeologous sequence variants, HSVs)被定义为亚基因组之间相应的单核苷酸变异 (Somers et al., 2003)。旁系同源序列变异 (Paralogous sequence variants, PSVs) 则定义为从一个相同的祖先序列分化出来,并存在于同一基因组中的序列拷贝间的单核苷酸差异 (Fredman et al., 2004)。在多倍体上的遗传应用研究上,如遗传作图、遗传多样性分析、关联分析等,上述三种类型的核苷酸变异,仅SNP具有应用价值。对SNP的开发需要测序不同品系/品种并准确区分同源和部分同源染色体之间的单核苷酸变异。除非使用基因组特异标记,否则几乎所有SNP标记分析平台必须面对这样的事实:即在SNP分析过程中,部分同源基因组上的序列拷贝容易与同源序列一起被取样分析。在这种情况下,如果异源四倍体两个基因组同一位点均出现多态,其后代的分离模式将与二倍体的不同(AA,AB 和 BB),按照AAAA、AAAB、AABB、ABBB和BBBB五种类型进行分离。此外,当异源四倍体某一个体中的SNP与另一个体上的HSV位点重合时,两个体的杂交后代将无法产生正常的等位分离模式,例如CT(A基因组)TT(B基因组) × CC(A基因组)TT(A基因组)。
栽培花生(Arachis hypogaea L.)属闭花授粉异源四倍体(AABB, 2n = 4× = 40),作为全球干旱和半干旱地区主要的经济作物,是人类营养中油脂和蛋白的重要来源。研究表明栽培花生形成的时间很短,大约在3500年通过野生种A. duranensis (A genome, n = 10) 和A. ipaensis (B genome, n = 10)融合产生(Seijo et al., 2004)。由于这个物种遗传基础狭窄,导致其DNA多态性极低,使其分子标记的开发和基因组学资源的挖掘一直以来面临很大的挑战(Paterson et al., 2004)。当前花生上开发利用的标记以SSR为主。而关于花生上SNP标记的开发只有零星报道。最近,Zhou et al., (2014)利用ddRADseq (double-digest restriction-site-associated DNA sequencing) 技术为两个栽培花生品系及其166重装自交系(RIL)后代构建简化文库(reduced-representation libraries,RRLs),并从两个亲本间检测出53,257 个候选SNPs,其中14,663个在RIL群体中同样被检出,但最终只有1,765个在随后的遗传图谱构建中得到验证。显然,在缺乏参考基因组序列以及序列变异参数条件下,利用第二代测序技术开发SNP的假性率高。虽然利用第二代测序技术挖掘SNP可节省单个标记的开发成本,但一旦所开发的SNP假阳性率高,将显著增加SNP的应用难度及成本。因此,弄清SNP、HSV和PSV在花生基因组中的频率,据此制定合适的SNP鉴定方法比仓促进行大规模SNP开发更稳妥可靠。
本研究的目的是:(1)利用扩增子测序方法鉴定EST-SNPs和genomic-SNPs;(2)估算花生基因组中SNPs, HSVs和PSVs的频率;(3)估算我国南方花生品种SNP的分布频率。
1结果与分析
1.1 SNPs、HSVs和 PSVs的鉴定
分析12个花生品种和2个花生二倍体祖先的EST和GSS扩增子表明,约90%的EST和GSS能够成功扩增出PCR产物。由于EST序列对应的基因组序列可能包含内含子,导致部分基因组扩增片段超出预期长度。为保证测序质量及避免内含子的干扰,本研究中片段长度超出预期的EST扩增子不用于后续的测序分析。此外,为保证测序的特异性,琼脂糖凝胶电泳检测出现多条扩增带的扩增子也不用于进一步测序分析。最终从EST和GSS扩增子中各挑选100个用于测序分析。结果表明,82%的EST扩增子和77%的GSS扩增子被成功测序。来自花生二倍体祖先A. duranensis和A. ipaensis的扩增子多为纯化子,其中EST扩增子的纯化率(87.8%)高于GSS扩增子(75.3%)。相反,四倍体花生的扩增子多为杂合子,其中GSS扩增子的杂合率(76.6%)高于EST扩增子(62.2%)。
花生二倍体祖先出现杂合扩增子说明存在旁系同源序列,而由于四倍体花生同时存在A和B基因组,其杂合扩增子的序列组成将更加复杂,可能包含部分同源基因,也可能包含旁系同源基因。序列比对发现,大部分扩增片段来自A和B基因组(图 1),但有部分只来自其中一个基因组。图2说明序列EST-86在花生A基因组中出现,但在B基因组中缺失。旁系同源序列的干扰以及异源多倍体进化过程中基因组序列的重组与缺失(Adams et al., 2005)往往增加了单核苷酸变异类型的区分难度。但通过比较四倍体花生及其二倍体祖先的扩增片段序列,多数单核苷酸变异类型能够被区分出来。如图3所示,通过序列比对可推断序列EST-87在栽培种上的两个拷贝来自A基因组,因此可确定拷贝间的序列变异为PSV,并非HSV,此外还可确定栽培品种YY7和TSSLR在第213碱基处的SNP位于A基因组。
.png) 图1 EST-48 在四倍体花生及其二倍体祖先上的扩增子序列 |
.png) 图2 EST-86 在四倍体花生及其二倍体祖先上的扩增子序列 |
.png) 图3 EST-87 在四倍体花生及其二倍体祖先上的扩增子序列 |
1.2 SNPs、HSVs和 PSVs频率
82个EST扩增子在12个花生品种中只有12个产生多态(至少包含1个SNP),共检测出18个EST-SNP,平均2557 bp出现一个EST-SNP。C/T (G/A)和A/C (T/G) (均占38.9%)为EST-SNP中最丰富的核苷酸变异类型,其次是A/T (T/A)和C/G (G/C),分别占16.7%和5.5%。77个GSS扩增子,有20个产生多态,共产生44个genomic-SNPs。genomic-SNP出现频率为1 SNP/1011 bp,远高于EST-SNP。genomic-SNP的核苷酸变异类型与EST-SNP相似,C/T (G/A)和A/C (T/G)最多(分别占68.2%和22.7%),其次是A/T (T/A)和C/G (G/C),分别为6.8%和2.3%。
1.3等位基因频率
本研究中共有32个包含SNP的扩增子可用于分析96个南方花生品种的多态性,其中30个扩增子只需分析1个SNP就足以对整个片段进行基因分型,因为扩增子包含的SNP均处于连锁不平衡状态。另外两个扩增子GSS-56和GSS-67,各包含2个连锁平衡的独立SNP。最终共有34个SNP可用于计算PIC值。对34个SNP位点设计巢式PCR引物,随后进行HRM分析。结果表明,除了GSS-35,其余33个扩增产物溶解曲线能够很好对调查材料进行基因分型(图4),其基因分型结果与前面的测序结果相吻合。SNPs是典型的双等位基因标记,因此单个SNP的PIC值不可能超过0.5。本研究中EST-SNP的PIC值介于0.021~0.413,平均为0.172(表1)。由于供试材料中只有一个品种在EST-12位点上产生等位变异,因此EST-12的PIC值仅为0.021。Genomic-SNP的PIC值显著高于EST-SNP,最低为0.08,最高为0.478,平均为0.249。
.png) 图4 扩增子 EST-66 的溶解曲线分析 |
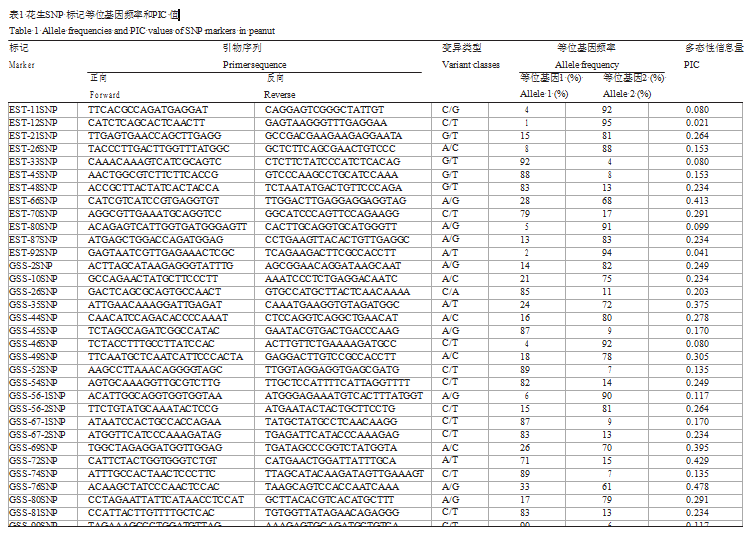 表1 花生 SNP 标记等位基因频率和 PIC 值 |
2讨论
本研究以四倍体花生及其二倍体祖先为材料,通过重测序EST和GSS开发SNP。如何鉴定出可真正用于基因分型的可靠SNP是当前多倍体SNP开发的最大挑战。为确保所开发SNP的可信度,首先必须将其与HSV和PSV区分开。这对于通过比较来自不同品种的EST或GSS直接开发SNP在生物信息学上无疑是一个难题。首先需要每个品种有大量高度冗余的序列,而目前NCBI数据库上面提供的花生EST或GSS序列尚未达到此测序深度。最近,Zhou et al. (2014)通过利用Illumina第二代测序平台大量平行测序四倍体花生基因组解决上述问题,共开发了53 257个候选SNP,但仅有极小部分在后续的遗传图谱构建中得到验证。因此,尚不清楚该研究中多大比例的SNP为假阳性。其他复杂的高通量测序技术的境遇也可能于此相同。
本研究显示花生EST-SNP和Genomic-SNP频率分别为1 SNP/2557bp和1 SNP/1011bp,显著低于其他自花授粉作物,如大豆、小麦、水稻、油菜。与此同时,花生HSV与PSV的频率则显著高于SNP,是其几十倍。因此,在序列拼接时,一旦将部分同源序列或旁系同源序列误判为同源序列,将显著增加SNP的假阳性率。因此,利用第二代测序技术开发SNP,适当提高序列的拼接标准,将有助于防止部分同源基因和旁系同源基因与同源基因错误拼接,降低SNP的假阳性率。
本研究所采用的扩增子测序是一个复杂的方法,其中光测序每个个体的扩增子就需要很大的工作量。针对异源多倍体植物,此法的优点是只有基因组上特异的序列才被用于后续的测序分析。在一个或两个基因组中扩增出多个位点的扩增子测序往往失败,因为扩增片段之间非常有可能出现插入缺失导致无法读取测序峰图。相反,只来自基因组内一个位点的扩增片段通常能被高质量测序。因此,结合二倍体祖先,通过对四倍体花生品种扩增产物的测序,最终能够开发出可靠的SNP。然而,由于花生基因组SNP发生频率偏低,单独采用扩增子测序存在低效率的问题。为兼顾SNP开发的效率和准确性,将第二代测序技术与扩增子测序相结合不失为SNP鉴定的好方法。首先利用第二代测序技术筛选大量的候选SNP,然后利用扩增子测序进行验证。
过去十年,新一代荧光双链DNA染料直接推动了HRM技术的发展(Wittwer et al., 2003)。新染料例如LCGreen和CYTO®9等具有毒性低,能够高浓度饱和双链DNA等优点。相比非饱和染料,新染料对PCR的抑制性低,与DNA结合力更强,熔解曲线分辨率更高(Wittwer et al., 2003; Monis et al., 2005),最终能检测出扩增产物中的SNP,甚至能发现体细胞突变和甲基化(Vossen et al., 2009; Mastoraki et al., 2015; Chang et al., 2014; Kristensen et al., 2008)。目前HRM在植物上的研究应用仍较少。最新的研究表明HRM是检测SNP、INDELS和SSR的有效工具(Chateigner-Boutin et al., 2007; Lehmensiek et al., 2008; Wu et al., 2008)。当溶解峰清晰明显时,HRM分析通常效果最好。为了使HRM分析到达更好的效果,需要充分考虑多个因素,包括(1)PCR产物长度和特异性;(2)GC 含量;(3)SNP 类型和密度(注意避开SNP 密集的区域);(4)PCR反应体系的优化等(Comai et al., 2006)。HRM分析能够检测到全部单碱基突变,其中对单核苷酸变异类型C/T (G/A)灵敏度高,而对A/T和G/C的灵敏度低。本研究中单核苷酸变异类型C/T (G/A)最为丰富,因此HRM适合用于检测花生SNP。
本研究表明花生genomic-SNP发生频率显著高于EST-SNP,几乎是EST-SNP的三倍,且genomic-SNP的PIC值也显著高于EST-SNP,说明genomic-SNP在花生基因组中分布广且均匀,因此,在进行遗传作图,关联分析等研究,GSS-SNP的效率将比EST-SNP高。
3材料与试剂
3.1植物材料
14个用于SNP鉴定的植物材料,包括(1)12个花生品种(表2);(2)2个花生野生种(二倍体祖先):A. duranensis (A基因组)和 A. ipaensis (B基因组)。96个南方花生被用于本研究中等位基因频率的调查,其中包含上述的12个品种(表2-1)。基因组DNA采用Moretzsohn et al. (2005)的方法从花生嫩叶中提取。
.png) 表2 96 个被调查的花生品种 |
3.2 PCR扩增
分别从Guo et al., (2008)开发的花生EST数据库和NCBI数据库(www.ncbi.nlm.nih.gov)中挑选EST(expressed sequence tags, EST)和GSS (Genome Survey Sequences)序列。采用Primer premier 5软件(Whitehead Institute for Biomedical Research, Cambridge, Mass)设计引物。引物设计基于下列原则:(1)溶解温度(Tm) 50℃~60℃,最适温度55℃;(2)扩增产物介于450 bp ~ 750 bp;(3) 引物长度18 bp至24 bp,扩增率大于80%;(4)引物GC含量40%~60%。PCR反应在PE9700热循环仪(美国ABI公司)上进行。PCR反应体系(20 μL):1×PCR buffer (50 mmol L-1 KCl,10 mmol L-1 Tris-HCl,pH8.3,1.5 mmol L-1MgCl2,0.1%明胶),1 U Taq DNA聚合酶,50~100 ng模板DNA,0.15 μmol L-1引物,0.2 mmol L-1 dNTP。反应条件为94℃ 5min;94℃ 1 min,55℃ 30 s,72℃ 1 min,35个循环;72℃ 5 min。单一条带PCR产物通过琼脂糖凝胶电泳检测确定。
3.3 Sanger测序和SNP挖掘
将12个花生品种和2个野生种的单一条带PCR产物送至北京六合华大基因科技股份有限公司,采用Sanger法进行双向测序。序列比对和SNP鉴定采用突变分析软件Mutation Surveyor®。
3.4 SNP基因分型
采用高分辨率溶解曲线(High Resolution Melting, HRM)方法对96个花生品种进行SNP基因分型。基于扩增子测序结果,在SNP侧翼序列设计用于HRM分析的巢式PCR引物。引物设计遵循以下原则:(1)扩增片段长度控制在80 bp至300 bp;(2)扩增片段内只包含一个SNP; (3)引物结合区含有HSV或PSV,以便设计亚基因组特异性引物。PCR反应容量为10 μl,除了在反应液中添加2.5 μM CYTO®9 (Invitrogen, Carlsbad, CA, USA) ,其余反应条件与上述相同。1 μl 100×第一次PCR产物稀释液作为PCR反应的DNA模板。
3.5统计分析
SNP标记的多态性信息量计算采用公式:PIC=.png)
其中k代表等位基因总数量,p为某一既定位点第i个等位基因的频率。
作者贡献
刘颖和朱方何负责扩增子测序;李杏瑜、洪彦彬和刘洪负责HRM分析;李杏瑜完成数据分析和论文初稿的写作;陈小平和李海芬参与论文修改;梁炫强是项目的构思者及负责人,指导实验设计,数据分析,论文写作与修改。全体作者都阅读并同意最终的文本。
致谢
本研究由现代农业产业技术体系建设专项(CARS-14)、广州市珠江科技新星专项(2013J2200088)、国家自然科学基金(31271767)、广东省自然科学基金(S2013020012647)、国家科技支撑计划(2013BAD01B05-4),广东省科技厅团队项目(2011A020102010)共同资助。
Anderson J.A., Churchill G.A., Autrique J.E., Tanksley S.D., and Sorrells M.E., 1993, Optimizing parental selection for genetic linkage maps, Genome, 36(1): 181-186
Botstein D., White R.L., Skolnik M., and Davis R.W., 1980, Construction of a genetic linkage map in man using restriction fragment length polymorphisms, Am. J. Hum. Genet., 32(3): 314-331
Chang C.C., Chang Y.S., Chan W.L., Yeh K.T., Wei R.J., and Chang J.G., 2014, Detection of SF3B3 gene mutations in oral cancer by high resolution melting analysis, Clin. Lab., 60 (12): 2023-2029
Chateigner-Boutin A.L., and Small I., 2007, A rapid high-throughput method for the detection and quantification of RNA editing based on high-resolution melting of amplicons, Nucleic Acids Res., 35(17): e114
Comai L., and Henikoff S., 2006, TILLING: practical single-nucleotide mutation discovery, Plant J., 45(4): 684-694
Fredman D., White S.J., Potter S., Eichler E.E., Den Dunnen J.T., and Brookes A.J., 2004, Complex SNP-related sequence variation in segmental genome duplications, Nat. Genet., 36 (8): 861-866
Guo B., Chen X., Dang P., Scully B.T., Liang X., Holbrook C. C., Yu J., and Culbreath A.K., 2008, Peanut gene expression profiling in developing seeds at different reproduction stages during Aspergillus parasiticus infection, BMC Dev. Biol., 8: 12
Kristensen L.S., Mikeska T., Krypuy M., and Dobrovic A., 2008, Sensitive melting analysis after real time-methylation specific PCR (SMARTMSP): high-throughput and probe-free quantitative DNA methylation detection, Nucleic Acids Res., 36 (7): e42
Lehmensiek A., Sutherland M.W., and McNamara R.B., 2008, The use of high resolution melting (HRM) to map single nucleotide polymorphism markers linked to a covered smut resistance gene in barley, Theor. Appl. Genet., 117(5): 721-728
Mastoraki S., Chimonidou M., Dimitrakopoulos L., Kounelis S., Malamos N., Georgoulias V., and Lianidou E., 2015, A rapid and accurate closed-tube methylation-sensitive high resolution melting analysis assay for the semi-quantitative determination of SOX17 promoter methylation in clinical samples, Clin. Chim. Acta, 444: 303-309
Monis P.T., Giglio S., and Saint C.P., 2005, Comparison of SYT09 and SYBR Green I for real-time polymerase chain reaction and investigation of the effect of dye concentration on amplification and DNA melting curve analysis, Anal. Biochem., 340(1): 24-34
Moretzsohn M.C., Leoi L., Proite K., Guimaraes P.M., Leal-Bertioli S.C., Gimenes M.A., Martins W.S., Valls J.F., Grattapaglia D., and Bertioli D.J., 2005, A microsatellite-based, gene-rich linkage map for the AA genome of Arachis (Fabaceae), Theor. Appl. Genet., 1(6): 1060-1071
Paterson A.H., Stalker H.T., Gallo-Meagher M., Burow M.D., Dwivedi S.L., Crouch J.H., and Mace E.S., 2004, Genomics and genetic enhancement of peanut, In: Wilson R.F., Stalker H.T., and Brummer C.E. (eds.), Genomics for legume crops, AOCS Press, Champaign, USA, pp.97-109
Seijo J.G., Lavia G.I., Fernández A., Krapovickas A., Ducasse D., and Moscone E.A., 2004, Physical mapping of the 5S and 18S~25S rRNA genes by FISH as evidence that Arachis duranensis and A. ipaensis are the wild diploid progenitors of A. hypogaea (Leguminosae),Am.J.Bot.,91(9):1294-1303
Somers D.J., Kirkpatrick R., Moniwa M., and Walsh A., 2003, Mining single-nucleotide polymorphisms from hexaploid wheat ESTs, Genome, 49(3): 431-437
Vossen R.H., Aten E., Roos A., den Dunnen J.T., 2009, High-resolution melting analysis (HRMA): more than just sequence variant screening, Hum. Mutat., 30(6): 860-866
Wittwer C.T., Reed G.H., Gundry C.N., Vandersteen J.G., Pryor R.J., 2003, High-resolution genotyping by amplicon melting analysis using LCGreen, Clin. Chem., 49(6 Pt 1): 853-860
Wu S.B., Wirthensohn M., Hunt P., Gibson J., and Sedgley M., 2008, High resolution melting analysis of almond SNPs de- rived from ESTs, Theor. Appl. Genet., 118(1): 1-14
Zhou X., Xia Y., Ren X., Chen Y., Huang L., Huang S., Liao B., Lei Y., Yan L., and Jiang H., 2014, Construction of a SNP-based genetic linkage map in cultivated peanut based on large scale marker development using next-generation double-digest restriction-site-associated DNA sequencing (ddRADseq), BMC Genomics, 15: 351