基于图像配准分析物种进化关系的新方法 
2中山大学生命科学学院, 广州, 510275
 作者
作者  通讯作者
通讯作者
计算分子生物学, 2012 年, 第 1 卷, 第 2 篇 doi: 10.5376/cmb.cn.2012.01.0002
收稿日期: 2012年03月12日 接受日期: 2012年06月30日 发表日期: 2012年07月10日
引用格式(中文):
严翠婷等, 2012, 基于图像配准分析物种进化关系的新方法, 计算分子生物学(online) Vol.1 No.2 pp.7-15 (doi: 10.5376/cmb.cn.2012.01.0002)
引用格式(英文):
Yan et al., 2012, A Novel Method for Evolution Analysis based on Image Registration, Jisuan Fenzi Shengwuxue (online) (Computational Molecular Biology) Vol.1 No.2 pp.7-15 (doi: 10.5376/cmb.cn.2012.01.0002)
图像配准是图像处理的一个重要技术,可用于分析两幅图像之间的相似度。本文提出了一种基于图像配准分析物种进化关系的新方法:首先利用一阶马尔可夫链方法计算不同基因组序列的寡聚核苷酸转移概率矩阵;然后将转移概率矩阵转换为彩色图像矩阵,并绘制物种两两之间彩色图像矩阵的联合直方图;最后分析联合直方图点集的分布情况,引入直方图点集的散度公式,将其作为相似性测度的标准,从而鉴定物种亲缘关系的远近。100种细菌全基因组的计算结果表明,相较于单基因法或基于基因组寡聚核苷酸频率组分差异信息的方法,本文提出的新方法具有更高的准确度和分辨力,它不仅能够很好地分辨科以下的分类单元,对科以上的分类单元同样具有较好的区分效果。该方法有望发展成为物种鉴定及系统发育推断的有效手段。
分析物种之间进化关系的传统方法是对同源基因进行多重序列比对(Wu and Eisen, 2008),但是,这种基于单个或多个基因的分析方法存在着局限性,其可靠性依赖于所选择的基因是否能够真实反映物种的进化历史。并且,该方法的分辨力有限,它只对种以上的分类单元具有较高的分辨力。据Mahoko等人的研究发现,在只利用16S rRNA基因构建物种的系统发育树时,种内菌株虽然能够正确聚类,但是彼此之间的系统发育关系却无法确定(Takahashi et al., 2009)。另一种方法则是基于物种的全基因组进行系统发育分析,它通常是比较物种基因组在GC含量或者寡聚核苷酸频率组分上的差异,进而分析物种之间亲缘关系的远近(Bohlin et al., 2008a; 2008b)。然而,这种方法只考虑了基因组中寡聚核苷酸的含量或相对丰度,却忽略了寡聚核苷酸的组成方式以及相邻寡聚核苷酸间的影响,因此也是片面的。而从适用性来看,熊远妍等(2008)人利用不同基因组中寡聚核苷酸频率组分差异的信息构建系统树,结果显示,该方法只有在分析科以下的分类单元时才能够得到比较合理的结果,而对科以上分类单元的分析结果则不理想。
鉴于此,我们提出了一个基于图像配准技术分析物种进化关系的新方法。为了充分考虑相邻寡聚核苷酸间的影响,在分析物种基因组时引入了马尔可夫链方法。我们假设基因组序列的延续是一个具有马尔科夫性质的离散时间随机过程,该过程中,序列中每一个寡聚核苷酸可以采取任何一种组合方式转移到下一个相邻的寡聚核苷酸,而这一步转移与之前的转移路径是无关的,其中与组合方式改变相关的概率叫做转移概率(Phillips et al., 1987)。利用一阶马尔可夫链方法分析基因组,可得到物种的寡聚核苷酸转移概率矩阵,该矩阵包含了基因组的全部信息,可用于推断物种的进化关系。接着在比较转移概率矩阵间的差异时,引入了图像配准技术(Pass and Zabih, 1999),通过绘制转移概率矩阵间的联合直方图,并采用联合直方图散度分析其点集的分布情况(梅跃松等, 2007)。相较于前面提到的传统方法,新方法的适用范围更广,准确度和分辨力更高,它不仅适用于种内亲缘关系十分接近的物种间的鉴定,还可区分目以上亲缘关系较远的物种。
1结果分析
1.1联合直方图散度可真实反映物种的进化距离
对表1中的100个物种全基因组进行分析,计算它们两两之间的联合直方图散度(HD),结果发现,对于大部分的物种,分类单元的级别越高,即物种之间的进化距离越大,物种的联合直方图散度也越大。例如,Streptococcus pneumoniae TIGR4 (简写为S. pneu_TI)与Streptococcus pneumoniae D39为同种的两个不同菌株,它们的HD为49.027 3;而S. pneu_TI与Streptococcus gordonii str. Challis substr. CH1为同属不同种的两菌株,它们的HD则为375.778 9;同样的,S. pneu_TI与Lactococcus lactis subsp. Cremoris MG1363、Lactococcus salivarius UCC118、Staphylococcus epidermidis ATCC RP62A、Clostridium botulinum F str. Langeland及Aster yellows withches’-broom phytoplasma AYWB分别为同科不同属,同目不同科,同纲不同目,同门不同纲及同界不同门的两菌株,它们的HD分别为1 488.71、2 015.58、2 339.58、3 054.30及4 088.10,呈现出一个递增的趋势。为了统计HD随着进化距离变化的总体趋势,按照“同种不同菌株”、“同属不同种”、“同科不同属”、“同目不同科”、“同纲不同目”、“同门不同纲”以及“同界不同门”的分类单元将所有物种两两之间的HD值分为7个小组,再计算各小组HD的平均值。各分组间进行两两t检验,组间显著性差异具有统计学意义(p<0.01)。其联合直方图散度按生物分类级别的分布呈现单调递增的趋势:即随着分类级别的升高,HD的平均值呈现出递增的趋势(图1)。我们的计算结果说明联合直方图散度在鉴定物种进化关系方面是一个可靠的指标,它能够真实地反映物种的进化距离。
|
|

|
|
.png)
1.2基于图像配准分析物种进化关系的新方法具有更高的分辨力
由于乳酸乳球菌与其他物种的种属关系比较清晰明了,在此以乳酸乳球菌为例对新方法的分辨力进行评估。例如Lactococcus lactis subsp. cremoris MG1363、Lactococcus lactis subsp. cremoris SK11以及Lactococcus lactis subsp. lactis Il1403 (分别简写为L. lac_MG, L. lac_SK与L. lac_I1)为乳酸乳球菌的3个不同菌株,其中Lla MG与Lla SK属于同一个亚种Lactococcus lactis subsp. Cremoris。一方面用欧几里徳距离公式计算三者之间的距离,结果显示,同亚种的L. lac_MG与L. lac_SK的欧几里徳距离为0.043 8;而不同亚种的L. lac_MG与L. lac_I1的欧几里徳距离为0.062 4 (表2),即同亚种与不同亚种这两个分类单元之间的差异仅为0.018 6。另一方面利用联合直方图散度比较上述三个菌株间的差异,结果发现,同亚种与不同亚种这两个分类单元之间的差异高达51.691 4 (表3)。该结果说明了联合直方图散度比欧几里徳距离更能清晰分辨种内亲缘关系十分接近的物种。
|
|
|
|
另外,将L. lac_MG与Pediococcus pentosaceus ATCC 25745、Staphylococcus aureus subsp. aureus USA300_FPR3757及Alkaliphilus metalliredigens QYMF (分别简写为P. pen_AT, S. aur_U及A. met_QY)进行比较,结果显示,L. lac_MG与同目不同科的P. pen_AT,同纲不同目的S. aur_US及同门不同纲的A.met_Qy间的欧几里徳距离分别为0.320 9、0.391 8及0.382 3,它们之间差异的平均值仅为0.047 3;而L. lac_MG与P. pen_AT,S. aur_US及A. met_Qy间的联合直方图散度分别为1 090.625 1,1 291.841 0及 2 305.814 0,它们之间差异的平均值为810.125 9,即联合直方图散度能够清晰分辨目以上亲缘关系比较远的物种。对金黄色葡萄球菌金黄亚种(Staphylo coccus aureus subsp. aureus)的6个菌株以及酿脓链球菌种(Streptococcus pyogenes)的6个菌株进行分析均能得到上述结论(表4; 表5 )。综上所述,联合直方图散度比欧几里徳距离在鉴定物种进化关系方面具有更高的分辨力。
|
|
|
|
1.3联合直方图散度可作为单基因系统发育树构建过程的补充
以葡萄球菌属中的5个不同菌株为例,其中有4个菌株均属于金黄色葡萄球菌,另一个属于腐生性葡萄球菌。基于16S rRNA的核苷酸序列构建它们的系统发育树,结果显示,同为金黄色葡萄球菌的4个菌株,虽然都能够聚类在一起,但是它们之间的系统发育关系却不明确(图2A),也就是说单基因树无法鉴定出种内菌株的进化关系。而基于物种全基因组三核苷酸转移概率矩阵的联合直方图散度,不仅能够将同种的4个菌株聚类在一起,还能够很好地分辨它们之间的系统发育关系(图2B)。因此,当单基因建树法无法分辨亲缘关系十分接近的物种时,可引入联合直方图散度这一参数,对其进行鉴定分析。换言之,联合直方图散度可作为单基因系统发育树构建过程的补充。在鉴定酿脓链球菌同种内的6个菌株时,同样能够得到上述结果(图3)。
|
|
.png)
|
|
.png)
2讨论
通过寡聚核苷酸转移概率矩阵分析物种间的进化关系,利用了全基因组的信息,能够真实地反映物种的进化历史。本文提出的新方法,基于图像配准技术,利用联合直方图比较物种之间的差异,并引入联合直方图散度这一参数度量此差异,所得结果具有更高的准确度及分辨力。无论是种以内的近缘物种,还是目以上的远缘物种,该新方法都能够有效地衡量它们之间的进化距离,这是因为联合直方图散度是基于对两个彩色图像矩阵间的灰度信息进行统计获得的,相较于单纯地计算寡聚核苷酸转移概率间的差值,新方法对物种基因组间的差异更为敏感,能更清晰地辨别不同物种间的进化距离。所以,这种基于图像配准分析物种进化关系的新方法,潜在有更广的适用性和更高的分辨力。
正如我们前面计算结果所证实的,对于大多数物种而言,联合直方图散度与物种间进化距离存在正相关关系:物种间的亲缘关系越远,它们之间的联合直方图散度就越大。然而,仍然存在某些特例,即个别亲缘关系较近的物种,其联合直方图散度反而大于亲缘关系较远的物种。究其原因,可能是微生物中基因水平转移的普遍存在,导致远缘物种间基因组的某些区段具有很高的相似性(Gogarten and Townsend, 2005)。另一个可能的原因是趋同进化现象的出现,导致不同的生物,甚至在进化上相距甚远的生物在基因组水平上产生了相似的变化(Amoutzias et al., 2004)。
基于图像配准分析物种进化关系的新方法不仅能够很好地分辨科以上的分类单元,与单基因建树法比较,对科以下的分类单元具有更好的区分效果。尽管目前这种用于物种进化分析的新方法还不够完善,对某些物种可能失效,但鉴于其对物种间差异的高度敏感性和分辨力,仍不失为物种鉴定及系统发育推断的一种有效手段和新型的辅助工具。例如可用于未知物种的鉴定(Tyagi et al., 2010),通过与已知进化谱系的物种进行联合直方图分析,计算它们的联合直方图散度,从而确定该未知物种所属的分类单元。下一步的研究计划是完善并合理利用这种方法,使之真正发展为一个行之有效的物种鉴定的新手段。
3材料与方法
3.1基因组数据
本论文中,用于计算联合直方图散度的100种原核生物全基因组序列均下载自NCBI (http://www.ncbi.nlm.nih.gov/sites/genome/)。这些物种的名称,NCBI登录号,分类单元ID号以及进化谱系等信息见表1。进化谱系的分类单元从门到种,并对各物种的进化谱系按照一定的规律进行简写。例如,这100个物种分别属于硬壁菌门(Firmicutes)、软壁菌门(Tenericutes)和变形菌门(Proteobacteria),可分别简写为F.、T.和P;而硬壁菌门下又包含了杆菌纲(Bacilli)和梭菌纲(Clostridia),又可分别简写为F.1和F.2,依此类推。因此,物种“Clostridium beijerinckii NCIMB 8052”的进化谱系为Firmicutes (Phylum1)-Clostridia (Class2)-Clostridiales (Order1)-Clostridiaceae (Family1)-Clostridium (Genus2)-Clostridium beijerinckii (Species2),可简写为F.2.1.1.2.2 (Sun et al., 2010; Qi et al., 2004)。
另外,在评估新方法中涉及到的11种原核生物16S rRNA核苷酸序列均下载自NCBI (http://www.ncbi.nlm.nih.gov/sites/gene/)。它们的Gene ID、Taxa ID以及进化谱系等信息见表6。
|
|
.png)
3.2利用马尔可夫链方法计算基因组寡聚核苷酸转移概率矩阵
对于每一个基因组,分别计算长度为n的各种寡聚核苷酸转移到下一个相邻的长度为n的寡聚核苷酸的频率。具体算法是分别以n或2n bp大小的滑动窗口,每次移动1 bp的方法统计每一种长度为n或2n的寡聚核苷酸出现的频数,然后根据公式(1)计算得到它们的转移概率。由于细菌基因组的顺义链与反义链均可编码蛋白质,为了全面统计物种基因组包含的信息,我们计算了两条链的寡聚核苷酸转移概率矩阵,并对二者进行加和。最终得到了一个 的转移概率矩阵。
寡聚核苷酸转移概率的计算公式:
.png) (1)
(1)
其中,f(ω1...ωn)为寡聚核苷酸ω1...ωn出现的频率;f(ω1...ωnωn+1...ω2n)为寡聚核苷酸ω1...ωnωn+1...ω2n出现的频率。Ρ(ωn+1...ω2n|ω1...ωn )表示在ω1...ωn存在的情况下,ωn+1...ω2n出现的概率,即ω1...ωn转移到ωn+1...ω2n的概率。
根据遗传信息传递过程的规律可知,基因组的转录、翻译都涉及到密码子的配对,而密码子是由三个单核苷酸组成的,每个密码子代表了一个氨基酸或者终止信号。为了能够将分析结果与遗传信息的传递过程结合起来,我们计算了每个物种基因组的三核苷酸转移概率矩阵,最终可分别得到一个64×64的转移概率矩阵。
3.3通过图像配准技术比较各基因组三核苷酸转移概率矩阵间的差异
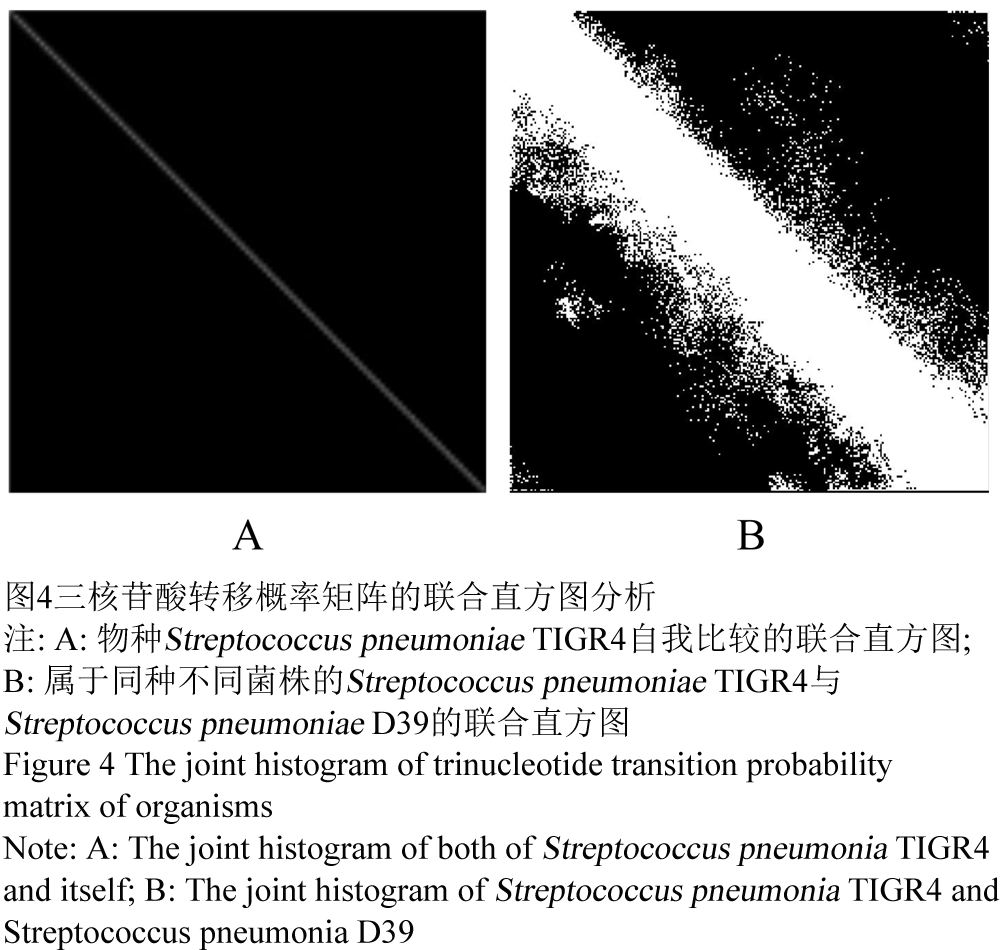
本文所采取的是基于灰度信息的图像配准方法。首先,将各物种基因组的三核苷酸转移概率矩阵转变为彩色图像矩阵。彩图矩阵中的每一个小方格对应着转移概率矩阵中相应位置的元素,根据元素值大小的不同,其对应小方格的颜色也不同(图4)。然后,通过联合直方图分析彩色图像矩阵间的差异。联合直方图在使用颜色信息的同时,还引入了两幅图像像素的位置信息,它实际上统计了两幅图像对应像素的不同灰度组合出现的频数。具体算法是先定义一个M×N的矩阵HIST [M, N],其中M和N分别为图4A和图4B的灰度级数。然后,对于每一个像素i∈A∩B,令HIST [A(i),B(i)]+1,其中A (i)和B (i)分别为图4A和图4B在像素i处的灰度。这样最终统计出来的HIST [M,N]就是图A和B的联合直方图矩阵(梅跃松等, 2007)。最后通过软件绘制出该联合直方图矩阵的图谱。因此,图A和图B越相似,它们的联合直方图谱就越趋近于45°线;反之,则以45°线为中心线,扩散程度越来越大(图5)。
|
|
|
|
.png)
由此可见,联合直方图的扩散程度可作为两幅图像的相似性测度,我们将之定义为联合直方图散度。换言之,联合直方图散度可作为推断物种间进化关系的标准,物种亲缘关系越接近,基因组的相似性越高,计算得到的联合直方图散度就越小,反之则越大。
联合直方图散度的计算公式:
.png) (2)
(2)其中,i、j分别代表矩阵HIST的行号和列号;HIST [i, j]代表矩阵HIST中第i行第j列的元素值。
公式(2)中,分子的几何意义是联合直方图上每一个点到45°线的距离平方加权和;分母的几何意义是两幅图像重合部分像素的数量,其作用是去除相似性测度与两幅图像重叠度的关联性(梅跃松等, 2007)。彩色图像矩阵的绘制和联合直方图散度的计算均通过软件MATLAB完成。
3.4基于图像配准分析物种进化关系方法的评估
依据一定长度的寡聚核苷酸组分差异计算序列间的距离,常用的是欧几里徳距离公式,即直接用组分差异的平方相加求和。为了评估基于图像配准新方法的分辨力,我们将物种间三核苷酸转移概率矩阵的欧几里徳距离与联合直方图散度进行比较,从而判断两者在表现序列间差异方面的优劣。
两个物种基因组寡聚核苷酸转移概率矩阵的欧几里徳距离计算公式:
.png) (3)
(3)其中,N为转移概率矩阵的总行数或总列数;xi, j、yi, j分别代表两个物种的转移概率矩阵中第i行第j列的元素值。
另外,基于单基因的系统发育分析方法存在着局限性且分辨力有限,它只对种以上的分类单元具有较高的分辨力,而对种以下亲缘关系十分接近的物种则难以区分(Bohlin et al., 2008)。对于用单基因序列比对无法区分的物种,我们用联合直方图散度进行聚类分析,由此评估联合直方图散度在系统发育分析方面的优势。这里,我们先用CLUSTAL X对11个物种的16S rRNA基因进行多重序列比对,接着用邻接法构建单基因系统发育树;同时,通过构建这些物种的联合直方图散度矩阵,并利用PHYLIP软件进行聚类,推断它们的系统发育关系;最后,通过比较上述两种手段构建的系统发育树,对新方法进行评估。
作者贡献
严翠婷是研究的主要执行人,包括数据采集、分析和初稿写作;黄庆生编写程序,参与部分数据分析和讨论;章芬参与部分数据分析;方颖是项目负责人,指导整个实验设计、数据分析、论文写作和修改。
参考文献
Amoutzias G.D., Robertson D.L., Oliver S.G., and Bornberg- Bauer E., 2004, Convergent evolution of gene networks by single-gene duplications in higher eukaryotes, EMBO Reports, 5(3): 274-279
http://dx.doi.org/10.1038/sj.embor.7400096 PMid:14968135 PMCid:1299007
Bohlin J., Skjerve E., and Ussery D.W., 2008a, Reliability and applications of statistical methods based on oligonucleotide frequencies in bacterial and archaeal genomes, BMC Genomics, 9(1): 104
http://dx.doi.org/10.1186/1471-2164-9-104PMid:18307761 PMCid:2289816
Bohlin J., Skjerve E., and Ussery W., 2008, Investigations of oligonucleotide usage variance within and between prokaryotes, PLoS Computational Biology, 4(4): 1-9
http://dx.doi.org/10.1371/journal.pcbi.1000057PMid:18421372 PMCid:2289840
Gogarten J.P., and Townsend J.P., 2005, Horizontal gene transfer, genome innovation and evolution, Nature Reviews Microb-iology, 3(9): 679-687
http://dx.doi.org/10.1038/nrmicro1204PMid:16138096
Mei Y.S., Yang S.X., and Mo B., 2007, Automatic image registration algorithm based on a novel similarity measu-rement, Yiqi Yibiao Xuebao (Chinese Journal of Scientific Instrument), 28(4): 336-339 (梅跃松, 杨树兴, 莫波, 2007, 一种基于新的相似性测度的自动图像配准算法, 仪器仪表学报, 28(4): 336-339)
Pass G., and Zabih R., 1999, Comparing images using joint histograms, Multimedia Systems, 7(3): 234-240
http://dx.doi.org/10.1007/s005300050125
Phillips G.J., Arnold J., and Robert I., 1987, Mono-through hexanu-cleotide composition of the Escherichia coli genome: A Markov chain analysis, Nucleic Acids Research, 15(6): 2611-2626
http://dx.doi.org/10.1093/nar/15.6.2611PMid:3550699 PMCid:340672
Qi J., Luo H., and Hao B.L., 2004, CVTree: A phylogenetic tree reconstruction tool based on whole genomes, Nucleic Acids Research, 32: W45-W47
http://dx.doi.org/10.1093/nar/gkh362 PMid:15215347 PMCid:441500
Sun J.D., Xu Z., and Hao B.L., 2010, Whole-genome based archaea phylogeny and taxonomy: A composition vector approach, Chinese Science Bulletin, 55(22): 2323-2328
http://dx.doi.org/10.1007/s11434-010-3008-8
Takahashi M., Kryukov K., and Naruya S., 2009, Estimation of bacterial species phylogeny through oligonucleotide frequency distances, Genomics, 93(6): 525-533
http://dx.doi.org/10.1016/j.ygeno.2009.01.009PMid:19442633
Tyagi A., Bag S.K., Shukla V., Roy S., and Tuli R., 2010, Oligonucleotide frequencies of barcoding loci can discriminate sp ecies across kingdoms, Plos One, 5(8): 1-9
http://dx.doi.org/10.1371/journal.pone.0012330PMid:20808837 PMCid:2924895
Wu M., and Eisen J.A., 2008, A simple, fast, and accurate method of phylogenomic inference, Genome Biology, 9: R151
http://dx.doi.org/10.1186/gb-2008-9-10-r151PMid:18851752 PMCid:2760878
Xiong Y.Y., Wang J.P., Lan Y.J., Wen M., and Zhang S.H., 2008, Evolutionary information of the diversity of oligonucleotide frequency of genomes, Zhongshan Daxue Xuebao (Zirankexue Ban) (Acta Scientiarum Naturalium Universitatis Sunyatsen), (2): 84-88 (熊远妍, 王军鹏, 蓝一杰, 文明, 张尚宏, 2008, 基因组寡聚核苷酸频率组分差异的进化信息, 中山大学学报: 自然科学版, (2): 84-88